



La insuficiencia suprarrenal secundaria aislada es una entidad poco frecuente, salvo en los casos asociados a tratamiento esteroideo prolongado1. En adultos se relaciona con traumatismos craneales o hipofisitis linfocítica de probable origen autoinmune. Presentamos el caso de una paciente con déficit parcial de hormona adrenocorticotropa (ACTH) asociado a esclerosis múltiple (EM), asociación no descrita previamente en la literatura. La EM es una enfermedad neurodegenerativa de gran heterogeneidad clínica producida por un proceso inflamatorio desmielinizante del sistema nervioso central de probable origen autoinmune, siendo la primera causa de discapacidad por enfermedad en adultos jóvenes.
Mujer de 39 años con historia de dos episodios de días de duración de focalidad neurológica, el último de ellos hace un año, por lo que había sido estudiada realizándose resonancia magnética cerebral y cervical que objetivó múltiples lesiones de sustancia blanca. Estos hallazgos eran compatibles con enfermedad desmielinizante, cumpliendo los criterios diagnósticos de EM2. Se inició en ese momento tratamiento con bolos de corticoides y, posteriormente, tratamiento con copolímero A.
Acude un año después a la consulta refiriendo mareos frecuentes, astenia e intolerancia al calor. Aporta una analítica realizada coincidiendo con dicha clínica que evidencia una glucemia basal de 48mg/dl. Afirma que los síntomas ceden con la ingesta y refiere que comenzaron previo al inicio del tratamiento con copolímero A. Actualmente no sigue ningún tratamiento. A la exploración física: talla 162cm, peso 57kg, IMC 21,7, tensión arterial 90/60 mmHg, sin otros datos de interés.
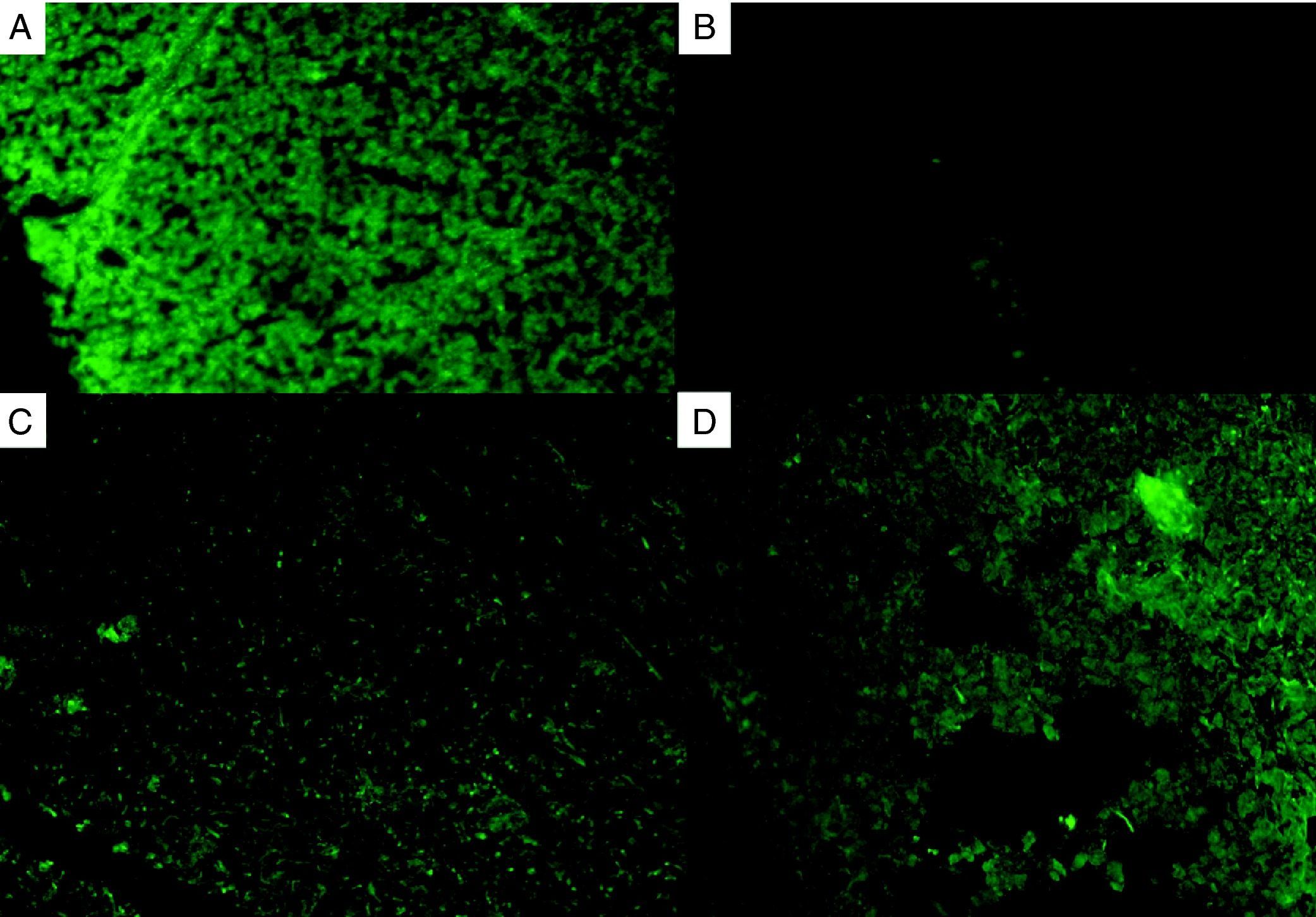
Las pruebas complementarias realizadas mostraron un hemograma y una bioquímica normales, glucemia 82mg/dL, insulina 7,67 mcUI/mL (9,3-29,1), HOMA 1,5, péptido C 1,8 ng/mL (1-1,5), TSH 1,3 mcUI/mL (0,3-5), T4L 1 ng/dL (0,8-1,7), IGF-1 195 ng/mL (109-284), ACTH 16 pg/mL (9-54), cortisol 3,3 mcg/dL (6,5-21), DHEA 166 mcg/dL (80-350). Se realizó la prueba de sobrecarga oral con 75g de glucosa que descartó la existencia de una pre-diabetes. Ante la sospecha de una insuficiencia suprarrenal, se realizó la prueba de estimulación con ACTH con respuesta en el límite bajo de la normalidad. Los anticuerpos anti-suprarrenal fueron negativos. Teniendo en cuenta el nivel de cortisol basal bajo con ACTH normal, se realizó una prueba de hipoglucemia insulínica para descartar una insuficiencia suprarrenal secundaria. Los resultados se exponen en la tabla 1. La respuesta de GH estaba dentro del rango de normalidad. En tiempo 60’ el cortisol basal respondió ligeramente, pero de forma global se observó que la ACTH y el cortisol apenas se modificaron tras la hipoglucemia intensa (34mg/dL a los 30 minutos) controlada con glucómetro. Se demostró, por tanto, una insuficiencia parcial de secreción de ACTH o insuficiencia suprarrenal secundaria parcial. Se realizó una nueva resonancia magnética, sin hallarse alteraciones a nivel hipofisario. Los anticuerpos antihipofisarios fueron positivos (fig. 1). Se inició tratamiento con hidroaltesona, inicialmente a 20mg/día repartido en tres dosis, con desaparición de la sintomatología. Por otra parte, no ha presentado nuevos brotes de EM tras un año de seguimiento.
Resultados de las pruebas funcionales.
| Sobrecarga oral de glucosa (75 g) | ||
| Glucemia (mg/dL) | Basal: 82 | A los 120 min: 67 |
| Estimulación con ACTH (Test de Synacthen®) | |||
| Basal | 30 min | 60 min | |
| Cortisol (mcg/dL) | 4,4 | 14,2 | 19,2 |
| ACTH (pg/mL) | 23 | – | |
| DHEA (mcg/dL) | 166 | ||
| Prueba de hipoglucemia insulínica | ||||||
| Basal | 15 min | 30 min | 45 min | 60 min | 90 min | |
| Cortisol (mcg/dL) | 5,0 | 4,2 | 4,1 | 7,2 | 12,8 | 7,8 |
| ACTH (pg/mL) | 20 | 19 | 19 | 38 | 27 | 22 |
| GH (ng/mL) | 0,3 | 0,2 | 0,3 | 8,2 | 8,7 | 3 |
| IGF-I (ng/mL) | 230 | |||||
| Glucemia | 85 | 34 | ||||
. A) Control positivo. B) Control negativo. C y D) Caso problema.")
Existen diferentes causas de hipoglucemia de ayuno en el paciente no diabético, entre ellas la insuficiencia suprarrenal y el déficit de hormona de crecimiento, por lo que deben incluirse dentro del diagnóstico diferencial. La insuficiencia suprarrenal secundaria aislada en el adulto se atribuye a etiología autoinmune en los casos en que se ha descartado su origen traumático. Suele debutar con síntomas inespecíficos como astenia, anorexia, pérdida de peso y tendencia a la hipoglucemia. Se ha publicado su asociación a otras enfermedades como infertilidad primaria, enfermedad de Crohn, miastenia gravis, poliquistosis renal, ataxia espinocerebelosa tipo 3 e hipertensión intracraneal idiopática, la mayoría de ellas de probable origen autoinmune3.
El cortisol plasmático basal es la prueba de elección como despistaje de insuficiencia suprarrenal; cuando es > 18 mcg/dL se excluye el diagnóstico. Con valores entre 3 y 8 es necesario realizar el test de estimulación con 250μg de ACTH, que sería normal si el cortisol a los 30 minutos es > 21, límite algo mayor que para descartar la insuficiencia primaria. Sin embargo, una respuesta normal no excluiría el diagnóstico de insuficiencia suprarrenal secundaria. El test de la hipoglucemia insulínica es la prueba más fiable para el diagnóstico de insuficiencia suprarrenal secundaria, siendo además el mejor predictor de la capacidad de secreción de ACTH en respuesta al estrés1. El tratamiento se basa en la sustitución con glucocorticoides, controlando de forma fundamentalmente clínica la respuesta al tratamiento.
La participación del eje hipotálamo-pituitario-adrenal (HPA) ha sido estudiada en enfermedades autoinmunes, entre ellas en la EM. Mason et al4 compararon ratas con baja respuesta del eje HPA al estrés con otras con respuesta normal, objetivando que las primeras eran más susceptibles a la encefalitis alérgica experimental, modelo in vitro la desmielinización por mecanismo inmune. Asimismo, también se constató experimentalmente la importancia de la activación del eje para la recuperación tras la desmielinización. Por tanto, en un principio se planteó la posibilidad de que los pacientes con EM pudieran tener una hipoactividad del eje que les haría más susceptibles a padecer la enfermedad. Sin embargo, en estudios posteriores se ha hallado una activación crónica del eje5; incluso se ha planteado su implicación pronóstica, de modo que una mayor hiperactivación se asocia a las formas de peor pronóstico6. En las placas desmielinizantes podría producirse una descarga de sustancias inflamatorias que actuarían sobre las neuronas que regulan la producción de CRH, influyendo secundariamente en el resto del eje. Otra posible explicación sería la afectación hipotalámica existente en la EM, como se ha demostrado en estudios anatomopatológicos7, o por la interrupción o la desmielinización de las vías relacionadas con la regulación de CRH. No obstante, si bien se ha objetivado que los pacientes con EM muestran una mayor activación de las neuronas productoras de CRH comparado con controles sanos, las lesiones activas hipotalámicas, en cambio, se correlacionan con una menor activación de las neuronas CRH e hipoactividad del eje, lo que iría en contra de esta última hipótesis8.
A pesar de que el origen del déficit aislado de ACTH es en muchos casos desconocido, se considera de mecanismo autoinmune. En nuestra paciente, la presencia de otra enfermedad autoinmune nos hace plantear esta etiología como posible causante del déficit de ACTH. La positividad de los anticuerpos antihipofisarios parece ir a favor de esta hipótesis, aunque por el momento estos anticuerpos no se han considerado buenos marcadores de la enfermedad por su baja sensibilidad y especificidad diagnóstica, así como por su variabilidad según el momento de la enfermedad y el método diagnóstico empleado para su detección9. La asociación de la EM con las enfermedades autoinmunes más frecuentes (artritis reumatoide, enfermedad tiroidea autoinmune, miastenia gravis, psoriasis) es bien conocida. La causa farmacológica en relación con el tratamiento con copolímero A es poco probable, dado que no cumple el criterio de temporalidad, habiendo aparecido los síntomas antes del inicio de este tratamiento. Por otra parte, exceptuando los tres bolos de metilprednisolona recibidos el año previo durante el brote de EM, la paciente no recibió tratamiento esteroideo por vía oral ni tópica. Otra posibilidad sería la supresión del eje secundaria a la afectación difusa del sistema nervioso central existente en la EM, bien por un mecanismo lesional de estructuras clave involucradas en el eje, o bien por un mecanismo inflamatorio desde las placas de desmielinización. En este sentido, los estudios realizados5,10 ponen de relieve la existencia de alteraciones del eje HPA en los pacientes con esclerosis múltiple que podrían ser relevantes en al menos un subgrupo de pacientes y que apoyarían la última hipótesis.
