


Neonato de 2 meses de edad diagnosticado de diabetes en el que se inició cetoacidosis con anticuerpos contra antígenos del islote negativos. En el estudio genético, se detectó la mutación R201C en el gen KCNJ11. En los últimos años se ha estudiado el cuadro de diabetes neonatal permanente debido a una mutación del gen KCNJ11 que codifica la subunidad Kir6.2 del canal del potasio sensible al adenosintrifosfato (KATP). Las mutaciones de Kir6.2 producen en última instancia una hiperpolarización de la membrana y la imposibilidad de secretar insulina al mantenerse los KATP permanentemente abiertos. Estudios recientes demuestran la efectividad de las sulfonilureas en el tratamiento. Las sulfonilureas se unen a la subunidad del receptor de sulfonilureas SUR1 y cierran el canal de forma independiente del ATP, de este modo restauran la secreción de insulina. Esta indicación no está aprobada debido a la falta de estudios de seguridad a largo plazo en lactantes; sin embargo, nos hace reflexionar sobre la importancia de la genética en la etiología y las implicaciones en su tratamiento.
A 2-month-old newborn was diagnosed with diabetes mellitus presenting with ketoacidosis and negative islet antibodies. Genetic study revealed the R201C mutation of the KCNJ11 gene. In the last few years, the heterozygous activating mutation in KCNJ11 encoding the Kir6.2 subunit of the ATP-sensitive potassium (KATP) channel has been shown to cause permanent neonatal diabetes. Diabetes results from impaired insulin secretion caused by failure of the beta cell-KATP channel to close in response to increased intracellular ATP. Recent studies have demonstrated the effectiveness of oral sulfonylurea in the treatment of this disease. Sulfonylurea closes the KATP channel by an ATP-independent route. Treatment with sulfonylurea in permanent neonatal diabetes has not yet been approved due to the lack of long-term studies in infants. However, the present case illustrates the importance of genetics to identify patients who may benefit from treatment.
La diabetes mellitus neonatal es una entidad poco frecuente que afecta a 1 de cada 400.000 recién nacidos1. Se define como la aparición de hiperglucemia antes del tercer mes de vida1–5. Existen dos formas: una transitoria con resolución en las primeras 12 semanas de vida y que precisa tratamiento insulínico al menos durante 2 semanas, relacionada con una anormalidad de la región cromosómica 6q24, y una forma permanente que requiere tratamiento con insulina de por vida1–5. Se sabe en la actualidad que la mutación en el gen KCNJ11 es la causa de más de la mitad de los casos de diabetes neonatal permanente1–6. Este gen codifica Kir6.2, subunidad del poro del canal de K+ sensible al adenosintrifosfato (Katp)7,8. Otra causa de diabetes neonatal permanente, descrita muy recientemente por Babenko et al9, tiene relación con la mutación en el gen ABCC8, que codifica el receptor de sulfonilureas (SUR1), la otra subunidad del canal KATP Además existen otras causas conocidas de diabetes neonatal permanente, como las mutaciones homocigotas de la glucocinasa y la aplasia pancreática debida a mutaciones del factor promotor de la insulina-1 (IPF-1)3,5,6,10–13.
La presentación clínica más habitual es una marcada hiperglucemia y un bajo peso al nacer debido a la disminución o ausencia de secreción de insulina intrauterina3,5,14, y la cetoacidosis es infrecuente. Los anticuerpos ICA (islet cell antibodies), anti-IA2 (proteína parecida a tirosinfosfatasa) y anti-GAD (descarboxilasa del ácido glutámico) son negativos y la concentración de péptido C está disminuida2,3,5,6. Se ha descrito algunas mutaciones con afección neurológica que combina retraso del desarrollo, debilidad muscular y epilepsia. Esta tríada constituye el síndrome DEND (delay, epilepsy and neonatal diabetes)1,4,10.
CASO CLÍNICONiña de 2 meses de edad que ingresó en la unidad pediátrica de cuidados intensivos por cetoacidosis diabética severa. La paciente nació, tras 42 semanas de gestación, por cesárea con un peso al nacimiento de 2.650 g (< percentil 10). Se trataba de la segunda hija de padres sanos, y sin antecedentes familiares de interés. Presentó al ingreso: glucemia capilar, 800 mg/dl; pH, 6,92; bicarbonato, 5,8; pCO2, 27,7; además, cetonuria, con glucohemoglobina (HbA1C) del 9,4%. Los anticuerpos anti-IA2, anti-GAD e ICA fueron negativos. Tras sueroterapia e insulinoterapia intravenosa intensiva a dosis iniciales de hasta 2 UI/kg/día, remitió el cuadro de cetoacidosis. Posteriormente inició lactancia materna y tratamiento con insulina lispro subcutánea después de cada toma y NFH nocturna, con una dosis total de 1 UI/kg/día. La HbA1C fue el 5,8% tras 4 meses de tratamiento y presentaba un percentil 25 de peso y longitud, con un desarrollo neurológico normal para su edad. En el estudio genético se detectó la mutación R201C en KCNJ11, de la que ninguno de los progenitores era portador.
DISCUSIÓNGloyn et al12 fueron los primeros que asociaron la diabetes neonatal con la mutación de Kir6.2, de tal modo que mutaciones heterocigotas de Kir6.2 aparecen en un 40-64% de los pacientes con diabetes neonatal permanente2. La mayoría de las mutaciones son de novo (> 90%). Las mutaciones más prevalentes son R201H, R201C, Y330C y V59M2,3,10–12.
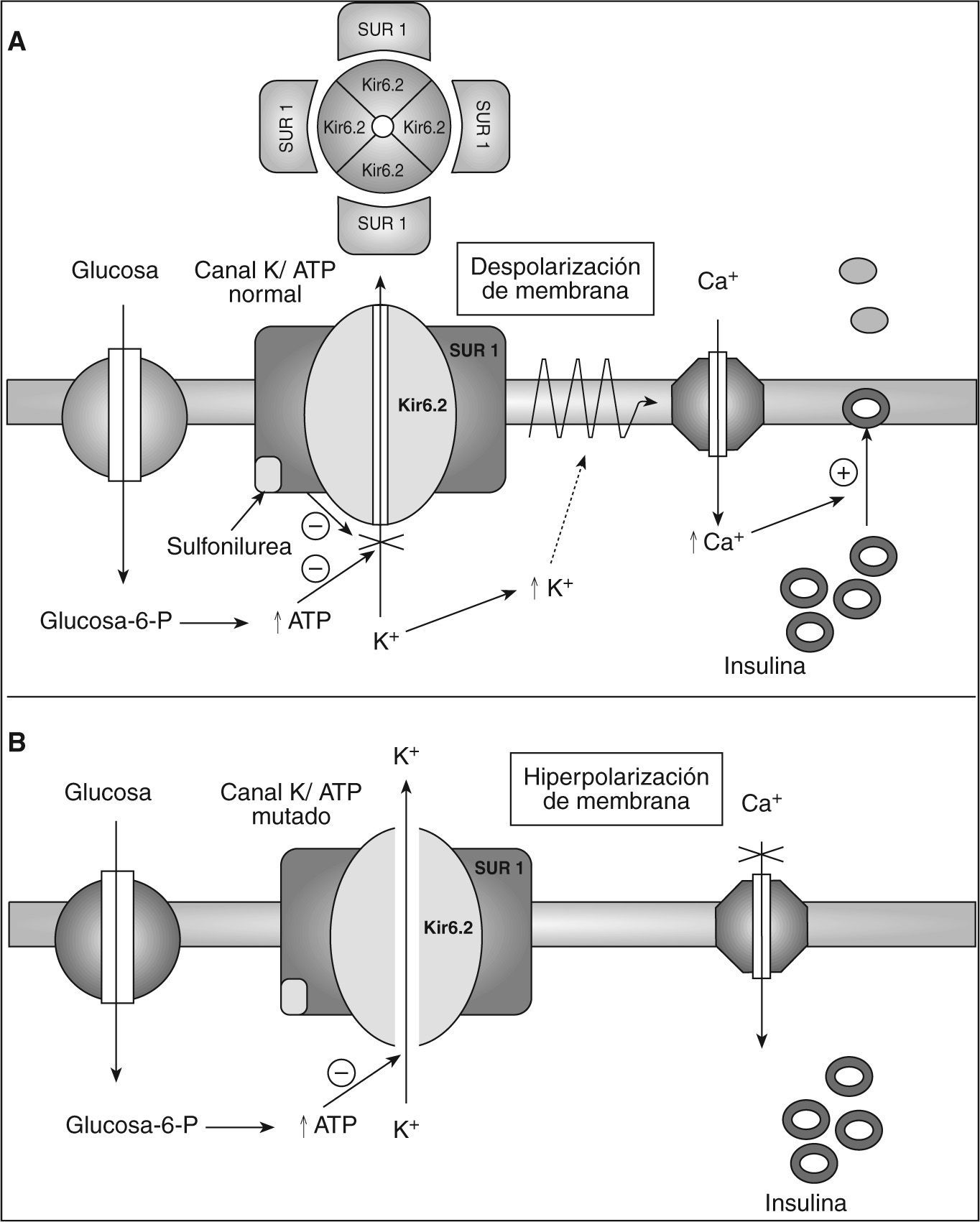
La importancia fisiológica de los KATP para la secreción de insulina es conocida desde hace muchos años7,8. Los KATP pertenecen a la superfamilia de los canales rectificadores internos del K+ (Kir), constituidos por cuatro subunidades Kir, llamadas Kir6.2 y 4 SUR1. Tanto Kir6.2 como SUR1 son necesarios para la correcta regulación metabólica del canal. La elevación de las concentraciones de glucosa activa el metabolismo de la célula beta y produce cambios en las concentraciones citosólicas de nucleótidos, el adenosintrifosfato (ATP) cierra el canal al unirse a Kir6.2, y los nucleótidos de magnesio (Mg-ADP y Mg-ATP) estimulan la actividad del canal interactuando con SUR12,4,13. Esto lleva a la despolarización de la membrana y a la posterior apertura de los canales de calcio dependientes de voltaje. El aumento de flujo de calcio al interior produce la exocitosis de los gránulos de insulina2,4,13 (fig. 1).
 en la célula beta y el mecanismo de acción de las sulfonilureas. B: consecuencias de la mutación de la subunidad Kir6.2 en los canales de potasio dependientes de ATP en la secreción de insulina. Glucosa-6-P: glucosa 6 fosfato; Kir6.2: subunidad del poro del canal de K+ sensible a ATP; SUR1: receptor de sulfonilureas.")
Representación esquemática de la secreción insulínica. A: funcionamiento normal de los canales de potasio dependientes de adenosintrifosfato (ATP) en la célula beta y el mecanismo de acción de las sulfonilureas. B: consecuencias de la mutación de la subunidad Kir6.2 en los canales de potasio dependientes de ATP en la secreción de insulina.
Glucosa-6-P: glucosa 6 fosfato; Kir6.2: subunidad del poro del canal de K+ sensible a ATP; SUR1: receptor de sulfonilureas.
Todas las mutaciones estudiadas conllevan en última instancia una disminución marcada en la habilidad del ATP para bloquear el canal de KATP2,4,13. Esta disminución implica que el canal permanezca abierto, se hiperpolarice la membrana y suprima el influjo de calcio y de la secreción de insulina2,4,13. El mecanismo molecular por el que la mutación de Kir6.2 disminuye la sensibilidad a ATP varía entre las diferentes mutaciones2,4,13 (fig. 1).
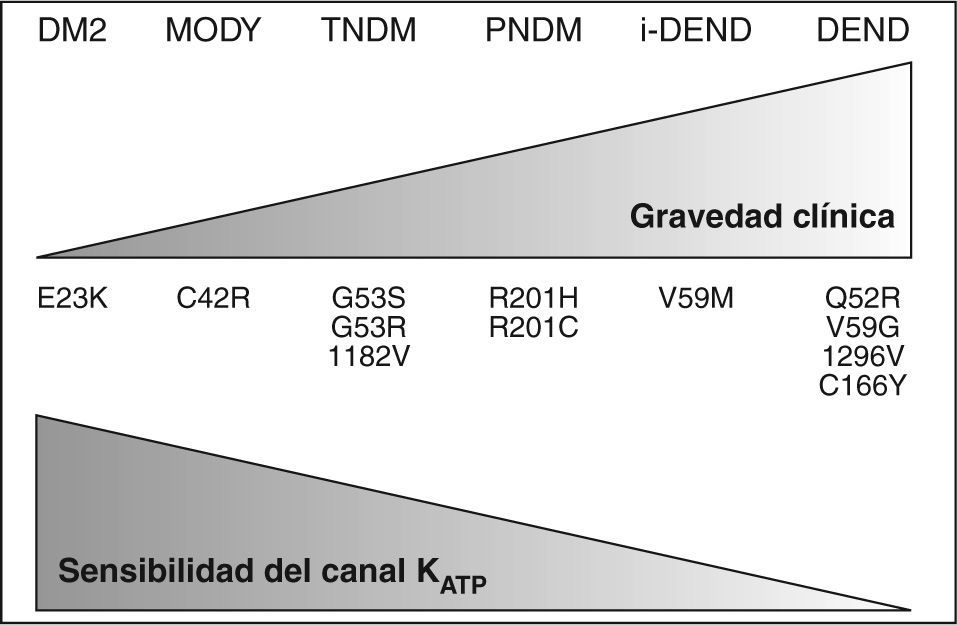
Hasta la actualidad se han descrito hasta 14 mutaciones15, con una presentación clínica variable en función del tipo de mutación y el número de subunidades del poro afectas4,12. Los síntomas neurológicos encontrados en algunos casos de diabetes neonatal, característicos de las mutaciones Q52R, I126L, V59G, C166Y, G53D y V59M12, probablemente se deban a la actividad de los KATP en otros tejidos como el muscular y el nervioso16 (fig. 2).

Relación entre genotipo, fenotipo y grado de disfunción en las distintas mutaciones de Kir6.2 y sus polimorfismos.
DEND: síndrome con diabetes neonatal, retraso en desarrollo y epilepsia; DM2: diabetes mellitus tipo 2; i-DEND: síndrome DEND intermedio; MODY: maturity onset diabetes of the young; PNDM: diabetes neonatal permanente; TNDM: diabetes neonatal transitoria.
La identificación de la mutación en el gen que codifica la subunidad Kir6.2 del canal de potasio tiene importantes implicaciones terapéuticas. Gloyn et al2 en diversos estudios in vitro hallaron que el canal de potasio podía regularse por un mecanismo independiente del ATP. Estudios iniciales con animales de experimentación demostraron la respuesta al tratamiento con tolbutamida intravenosa, con restablecimiento de la secreción insulínica2. Posteriormente estudios clínicos con glibenclamida vía oral corroboraron dichos resultados10,16–19.
Las sulfonilureas se unen a SUR1 y cierran el KATP, lo que permite que se despolarice la membrana y se restablezca la funcionalidad de la célula beta, pudiendo responder a GLP1 y a otros secretagogos10,18,19 (fig. 1).
Un estudio reciente de Pearson et al20 incluyó a 49 pacientes con diabetes mellitus neonatal con edades comprendidas entre 3 meses y 36 años, procedentes de 40 familias con mutaciones en KCNJ11. Los pacientes fueron tratados con glibenclamida a una dosis media de 0,45 mg/kg (intervalo, 0,05-1,5 mg/kg); 44 de ellos pudieron dejar el tratamiento con insulina, 38 pacientes obtuvieron una mejoría en el control glucémico y se produjo una disminución significativa del 1,7% en la HbA1C tras 12 semanas de tratamiento, sin incremento del número de hipoglucemias. Se observó además un marcado aumento de la secreción de insulina en respuesta a la sobrecarga oral de glucosa. Los pacientes requirieron dosis altas de sulfonilureas comparado con la dosis máxima recomendada en adultos con diabetes mellitus tipo 2; sin embargo, como efecto adverso apareció solamente diarrea transitoria. Babenko et al9 también demostraron una respuesta adecuada al tratamiento con sulfonilureas en 5 de 9 pacientes con mutación de la subunidad SUR1 del KATP.
La efectividad del tratamiento depende del tipo de mutación del canal. En la serie de Pearson, 5 de 6 pacientes con la mutación R201C respondieron satisfactoriamente al tratamiento, a diferencia de otras mutaciones, como la R201H en la que respondieron 23 de 23 pacientes20.
Las mutaciones que afectan a la parte del receptor implicado en la apertura del canal, como aquellas con alteración neurológica, son menos sensibles a la inhibición por sulfonilureas y requieren dosis mayores4,15. Sin embargo, el tratamiento con sulfonilureas cerraría también los canales de K+ en otras partes y mejoraría los síntomas neurológicos4,15.
Con este caso se ilustra el principio farmacogenético aplicado a algunos pacientes diabéticos y cómo la definición genética de su etiología puede servir para elegir el tratamiento óptimo. La falta de aprobación del uso de sulfonilureas en lactantes dificulta su tratamiento. Futuros estudios evaluarán la indicación de estos fármacos como tratamiento de primera elección de la diabetes neonatal con mutación del gen KCJN11. En cualquier caso, se necesitan otros estudios a largo plazo que verfiquen la seguridad del fármaco.
