


Los paragangliomas (PGL) son tumores neuroendocrinos poco frecuentes (prevalencia 1/1.700) derivados de líneas celulares de la cresta neural y localizados en la médula suprarrenal (feocromocitomas [FEO]), quimiorreceptores y ganglios simpáticos y parasimpáticos1. La clínica de presentación depende de la localización, perfil secretor y potencial maligno. Aproximadamente el 25% de los FEO y PGL son de origen familiar y se encuentran incluidos en diferentes síndromes como el Von Hippel Lindau (VHL), neoplasia endocrina múltiple tipo 2 (MEN2), neurofibromatosis tipo 1 (NF1) y el síndrome PGL/FEO debido a mutaciones germinales de la enzima succinil deshigrogenasa (SDH)2, que participa en la transferencia electrónica y en el ciclo de Krebs y expresa gran heterogeneidad fenotípica3.
Presentamos el caso de un paciente con un síndrome paraganglioma/feocromocitoma cuyo diagnóstico se realizó 25 años tras los primeros signos de la enfermedad.
Varón de 65 años de edad que ingresa por estreñimiento y dolor abdominal de 15 días de evolución refiriendo síndrome constitucional desde hacía 6 meses. Se realizó endoscopia preferente, objetivándose una masa estenosante en la unión recto-sigma requiriendo colocación de prótesis de Wallflex sin posibilidad de realizar biopsia.
Antecedentes personales: diabetes mellitus tipo 2, hipertensión arterial y enfermedad pulmonar obstructiva crónica (EPOC). Diagnosticado en 1985 de paragangliomas carotídeos bilaterales tratados mediante resección quirúrgica y un paraganglioma timpánico derecho. En el curso de la enfermedad presentó dos recidivas que fueron tratadas con radioterapia y tomoradiocirugía. Antecedentes familiares: padre fallecido con tumoraciones cervicales. Hija de 30 años diagnosticada a los 15 de paraganglioma timpánico y recientemente de paraganglioma carotídeo.
En las determinaciones analíticas destacaba una glucemia de 169mg/dl y antígeno de carcinoma embrionario (CEA) de 10,7ng/ml (0-5ng/ml). Se realizó TAC abdominopélvico que demostró una masa necrosada en retroperitoneo interaortocavo de 4,3cm, una masa suprarrenal izquierda de 2,6cm y otra en suprarrenal derecha de 1,5cm junto con una masa en la unión rectosigma.
En el interrogatorio dirigido, el paciente refería cefalea, sudoración, palpitaciones, temblor y nerviosismo prácticamente a diario desde hacía años que había sido etiquetado de síndrome ansioso-depresivo, en tratamiento crónico con lorazepam. Presentaba deterioro del estado general, disfagia, parálisis del nervio recurrente derecho, peso de 58kg y TA 105/58mmHg. En la evolución, presentó crisis de hipertensión arterial paroxística. No se objetivaron estrías, neurofibromas, hábito cushingoide, manchas café con leche ni nódulos de Lisch.
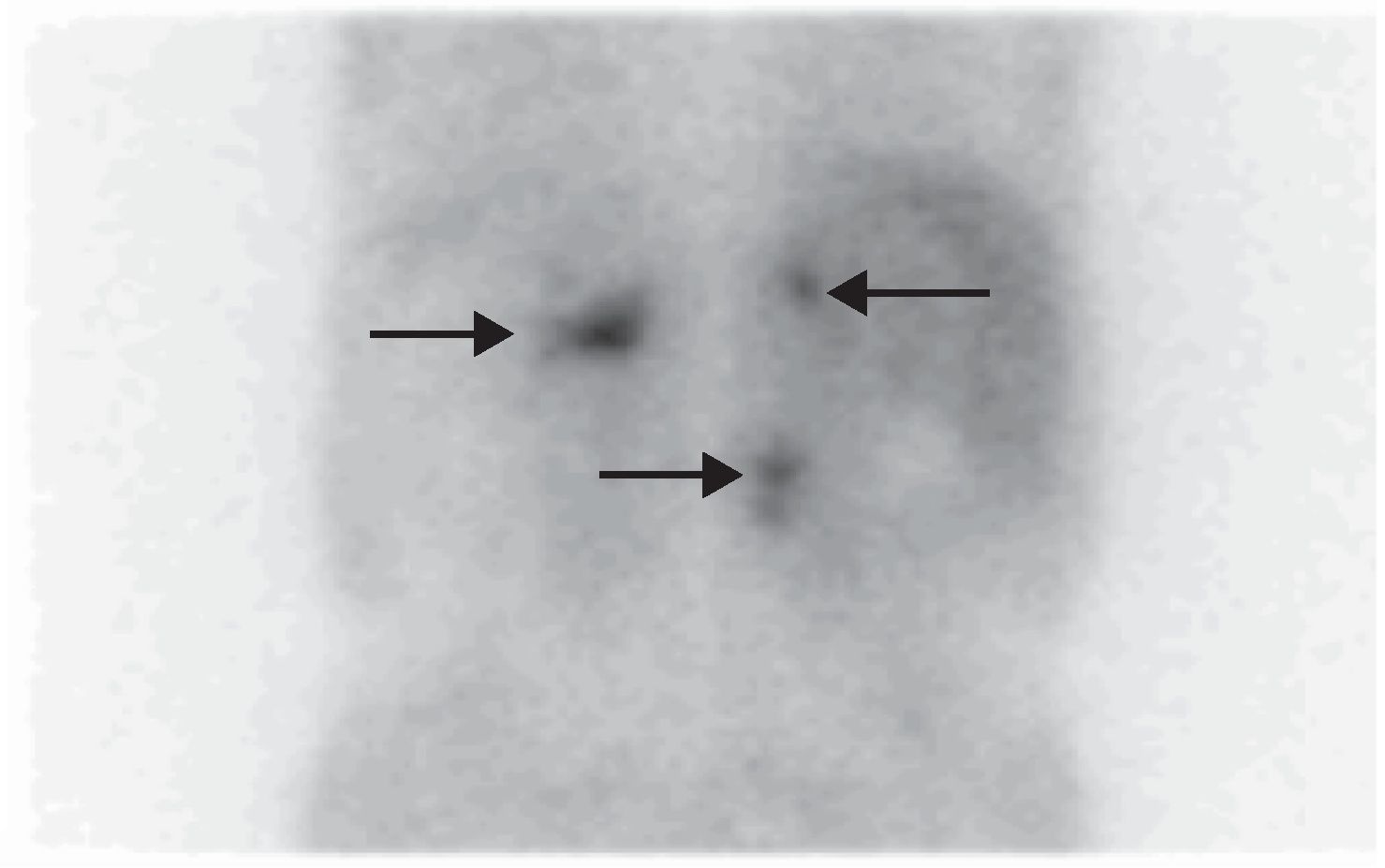
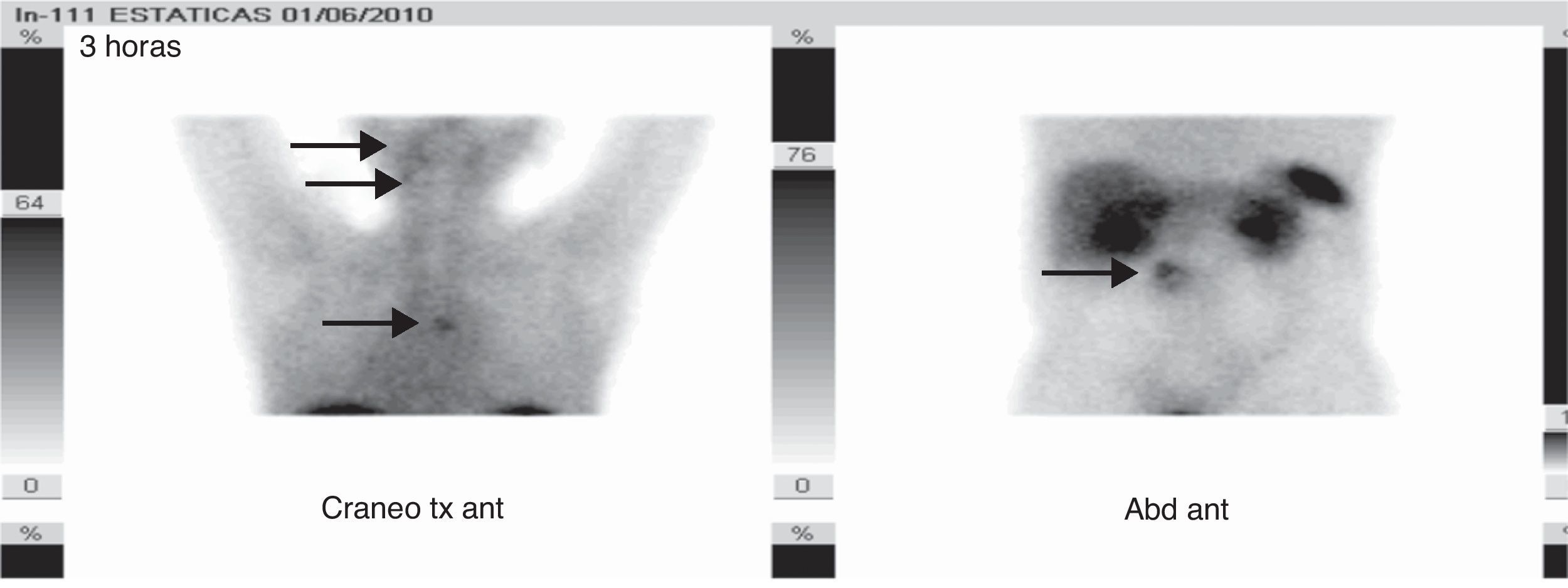
Ante los antecedentes familiares, la presencia de los paragangliomas de cabeza y cuello, la clínica del paciente y los nuevos hallazgos radiológicos, se consideró como primera posibilidad diagnóstica que los tumores suprarrenales fuesen feocromocitomas bilaterales funcionantes y el tumor retroperitoneal un nuevo paraganglioma abdominal, lo que englobaría todo el espectro clínico tumoral dentro de un mismo cuadro, el síndrome paraganglioma/feocromocitoma relacionado con la SDH. La neoplasia estenosante de rectosigma podría tratarse además de un tumor del estroma intestinal (tumor de GIST), que se asocia a dicho síndrome, aunque la localización del mismo era más compatible con un adenocarcinoma intestinal. En base a ello se realizaron determinaciones de catecolaminas y metanefrinas fraccionadas en orina de 24 horas (adrenalina<10μg/24h [0–25], dopamina 204μg/24h [0–400], normetanefrina>1.171μg/24h [0–500], noradrenalina 733μg/24h [0–80], metanefrina 55μg/24h [0–300], calcitonina<2pg/ml [0-18,2] y cromogranina A 1904,2ng/ml [19,4–48,1]). Se solicitó gammagrafía con metayodobencilguanidina[131I] que demostró intensa hipercaptación en las lesiones de ambas suprarrenales y de la masa retroperitoneal (fig. 1); gammagrafía de extensión tumoral con radiotrazador[111In]octeotride que mostró hipercapatación a nivel timpánico derecho, carotídeo derecho, mediastínico y paravertebral lumbar (fig. 2); y estudio genético para la mutación de la enzima SDH, que reveló la mutación c.197G≥A/p.W66X en heterocigosis en el exón 2 del gen 11q23 de la SDH tipo D, que consiste en un cambio de guanina por adenina en el nucleótido 197 del cDNA. Mutación previamente descrita y demostrada su asociación con la enfermedad4.


Tras preparación quirúrgica con bloqueo α adrenérgico con fenoxibenzamina se realizó exéresis quirúrgica de ambas suprarrenales, de la masa retroperitoneal y del tumor de la unión rectosigma.
El estudio histológico reveló dos feocromocitomas de 9 y 19mm, un paraganglioma extraadrenal de 20mm y un adenocarcinoma de bajo grado que infiltraba grasa perirrectal, vasos linfáticos y tractos nerviosos (T3N2M0).
Dos meses tras la intervención, mantenía glucemias normales sin tratamiento, hipotensión arterial, metanefrinas en orina y cromogranina A en rango normal, persistiendo CEA elevado. Precisó soporte nutricional a través de gastrostomía endoscópica percutánea. El TAC abdominal mostraba dos lesiones hepáticas sugestivas de metástasis mientras la gammagrafía [111In]octeotride no revelaba captación a nivel hepático ni abdominal, persistiendo las captaciones previas a nivel mediastínico y yugulotimpánicas, lo que sugería, junto con los marcadores, la progresión del adenocarcinoma de colon por lo que el paciente fue remitido para seguimiento oncológico.
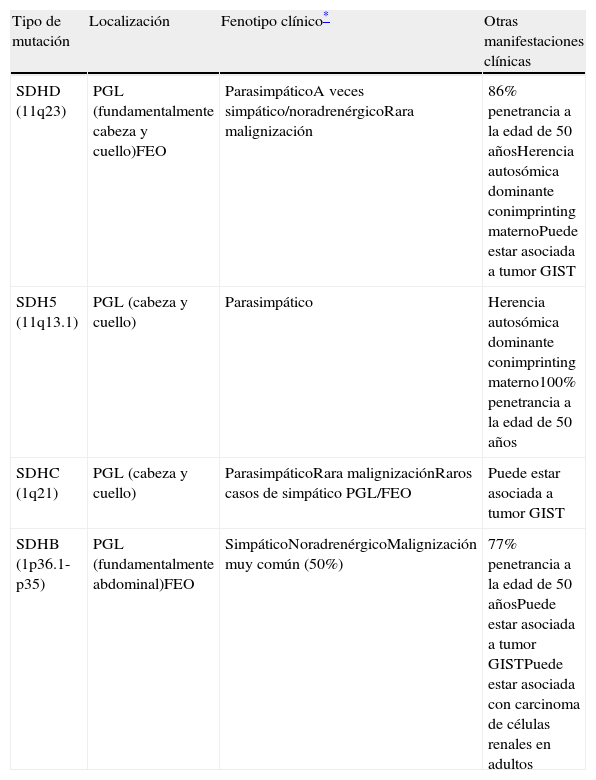
Los síndromes paragangliomas/feocromocitomas relacionados con la SDH se heredan con carácter autosómico dominante con penetrancia incompleta y dan lugar a múltiples paragangliomas de diferente localización. Esta enzima forma parte del complejo II del canal de la mitocondria respiratoria. Está compuesta por 4 subunidades que codifican 4 genes nucleares: SDHA, SDHB, SDHC y SDHD. Diferentes mutaciones en cada una de estas subunidades darán lugar a diferente genotipo-fenotipo y son responsables del 80% de las agregaciones familiares de los PGL y FEO5 (tabla 1). El resto de las enfermedades de carácter hereditario que pueden asociar feocromocitomas y paragangliomas son el MEN 2, el Von-Hippel-Lindau y la neurofibromatosis tipo 1 que en el paciente se descartaron, ya que asocian otras características reconocibles por exploración clínica y complementaria, que el paciente no presentaba.
Características de las mutaciones de la SDH
| Tipo de mutación | Localización | Fenotipo clínico* | Otras manifestaciones clínicas |
| SDHD (11q23) | PGL (fundamentalmente cabeza y cuello)FEO | ParasimpáticoA veces simpático/noradrenérgicoRara malignización | 86% penetrancia a la edad de 50 añosHerencia autosómica dominante conimprinting maternoPuede estar asociada a tumor GIST |
| SDH5 (11q13.1) | PGL (cabeza y cuello) | Parasimpático | Herencia autosómica dominante conimprinting materno100% penetrancia a la edad de 50 años |
| SDHC (1q21) | PGL (cabeza y cuello) | ParasimpáticoRara malignizaciónRaros casos de simpático PGL/FEO | Puede estar asociada a tumor GIST |
| SDHB (1p36.1-p35) | PGL (fundamentalmente abdominal)FEO | SimpáticoNoradrenérgicoMalignización muy común (50%) | 77% penetrancia a la edad de 50 añosPuede estar asociada a tumor GISTPuede estar asociada con carcinoma de células renales en adultos |
Los tumores simpáticos son funcionantes y secretan catecolaminas. Los tumores parasimpáticos son no funcionantes. En general, los paragangliomas de cabeza y cuello son parasimpáticos, mientras que los feocromocitomas y los paragangliomas abdominales son simpáticos. Los tumores noradrenérgicos secretan casi todos exclusivamente noradrenalina y normetanefrina, mientras que los tumores adrenérgicos secretan adrenalina y metanefrina además de nordrenalina y normetanefrina.
El curso de la enfermedad es relativamente benigno. La transmisión es exclusivamente paterna con «imprinting materno»: cuando la mutación se hereda de la madre, los hijos son portadores de la mutación pero no desarrollan la enfermedad.
La determinación de metanefrinas representa el mejor test bioquímico para diagnosticar feocromocitomas o paragangliomas funcionantes. Las metanefrinas plasmáticas libres o totales y las fraccionadas en orina de 24 horas presentan una sensibilidad (S) diagnóstica del 96, 95 y 95%, respectivamente con especificidades (E) del 89, 91 y 86%, repectivamente6.
El TAC y la RMN (S 98% y E 70%) son las pruebas de localización de primera elección, aunque la RMN parece presentar una mayor rentabilidad para los tumores de localización extraadrenal7.
La gammagrafía con metayodobencilguanidina [131I] MIBG sigue siendo de primera elección para la localización de los feocromocitomas secretores, ya que presenta una elevada S (94%) y E (92%) y permite confirmar la naturaleza secretora del tumor, localizar tumores no visibles en TC/RMN e identificar otros sitios de enfermedad8.
La gammagrafía de extensión tumoral con radiotrazador [111In] octeotride muestra en las series una S del 88-97% y E del 75-82%, sobre todo en pacientes con sospecha de enfermedad metastásica o extraadrenal.
Últimamente se han desarrollado nuevos radiotrazadores como la fluorodopamina [18F]-DOPA, la fluorodihidroxifenilalanina [18F]-FDA, la fluor-desoxiglucosa [18F]FDG o la dota-Nal octeotride DOTATOC. El valor diagnóstico de la [18F]-DOPA PET para la detección de los PGL extraadrenales no metastásicos parece superior al del [111 In]octeotride, con una sensibilidad del 95,7%, presentando una rentabilidad mayor sobre todo en los PGL≤10mm y en los abdominales. Los PGL de cabeza y cuello son más fácilmente reconocidos en ambas pruebas9.
Como en nuestro caso, para optimizar el diagnóstico de estos síndromes se deben utilizar varias pruebas de localización con carácter complementario.
