



The prevalence of chronic renal insufficiency and its complications, including dyslipidemia, is increasing. Although the characteristics of dyslipidemia in chronic renal insufficiency and its pathophysiology are well known, its cardiovascular and renal impact and the most effective therapeutic approach are poorly defined.
La prevalencia de insuficiencia renal crónica y sus complicaciones, como la dislipemia, ha ido en aumento. Aunque sabemos las características de la dislipidemia asociada a IRC y su fisiopatología, no está bien definido cuál es su impacto cardiovascular y renal y cuál es su mejor enfoque terapéutico.
Lipids are hydrophobic molecules with low solubility in water, while lipoproteins are a family of structurally similar particles1 consisting of large spherical complexes that function as transport vehicles for lipids in blood.1–4 These substances also carry fat-soluble vitamins, drugs, viruses, and antioxidant enzymes.5 They have a central hydrophobic core formed by triglycerides and esterified cholesterol and a more hydrophilic surface monolayer consisting of proteins, free cholesterol and phospholipids.2,3,6 High density lipoprotein (HDL) are the smallest and most dense lipoproteins, while the chylomicrons and very low-density lipoproteins (VLDL) are the largest and least dense. Their density is inversely related to size.3 Most triglycerides are transported in chylomicrons and VLDL, while most cholesterol is transported in the form of esterified cholesterol in low-density lipoprotein (LDL) and HDL.3,4 About 70% of total plasma cholesterol is in LDL.1,5
Apolipoproteins (apo) on the surface of lipoproteins are necessary for the assembly and structure of lipoproteins3 and are important regulators of enzyme activity or function as receptor ligands.1,4 There are five main classes from A to E.7 Lipoproteins containing apo-B, particularly their remnants [VLDL, intermediate-density lipoproteins (IDL), oxidized LDL, lipoprotein(a) (Lp(a)), remnants of chylomicrons], are considered atherogenic.5
Three main pathways are responsible for the generation and transport of lipids within the body. These pathways include the exogenous pathway, the endogenous pathway, and the pathway of reverse cholesterol transport. The exogenous transport pathway of dietary lipids3,4 essentially consists of the formation of chylomicrons, which carry dietary lipids absorbed from the intestine through the lymphatic system. In blood, circulating chylomicrons interact at the capillaries of adipose tissue and muscle cells releasing triglycerides to the adipose tissue to be stored and available for the body's energy needs. Through the endogenous pathway, the liver secretes triglyceride-rich VLDL, which transport these lipids from the liver to peripheral tissues2–5; the product of VLDL catabolism is intermediate density lipoprotein (IDL), which can be removed from plasma by the LDL receptor or undergo further processing, initially by lipoprotein lipase (LPL) and finally by hepatic lipase, resulting in LDL.1–3,5 Finally, by means of the reverse cholesterol transport pathway, HDL particles aid the redistribution of lipids between lipoproteins and the body's cells. This lipoprotein acquires cholesterol and transports it to the liver for excretion or to other cells that need it.3,4 The cardiovascular protective role of HDL may be related to the usefulness of the reverse cholesterol pathway, which leads this lipid away from the arterial wall and inhibits monocyte adhesion; through its antioxidant activity, this pathway prevents LDL oxidation. HDL contains paraoxonase,5,8 which has antioxidant activity, reducing oxidized LDL.8
DyslipidemiaDyslipidemia may be classified as primary, when it results from genetic defects that directly affect the metabolism of lipoproteins, or secondary, when it results from other disorders that indirectly affect the metabolism of lipoproteins, such as diabetes mellitus, hypothyroidism, sepsis, autoimmune diseases, different classes of drugs, liver disease, and chronic renal insufficiency (CRI), which will be discussed in this article.2,5 The recognition of secondary disturbances is of great importance, since treatment must be, at least partially, directed to the underlying disease.5
Dyslipidemia in chronic kidney diseaseChronic renal Insufficiency (CRI) is defined as a sustained and significant reduction1 (for more than 3 months) in the glomerular filtration rate (GFR)3 and may result from different conditions,1 which may be inherent to the kidney – primary renal disease – or a consequence of a systemic disease such as diabetes mellitus – secondary renal disease.6 Overall diabetes mellitus hypertension, focal segmental glomerulosclerosis, and autosomal dominant polycystic kidney disease account for 70% of all cases of end stage renal failure (ESRD) in the United States, while nonspecific glomerulonephritis accounts for 14%.1 Thus, with the increase of the elderly population and individuals with diabetes and hypertension, CRI is a growing health problem.9,10 At the same time there is an increase in the complications associated with CRI, including anemia, malnutrition, osteoarthritis, and dyslipidemia.9
The spectrum of dyslipidemia in patients with CRI is distinct from that in the general population.4,11 This disorder involves all classes of lipoproteins,4 occurs in all stages of CRI12 – including mild disease – in patients without supportive treatment, and in patients on dialysis or after kidney transplantation.6
In 1836 Richard Bright commented on the “milky serum” of patients with ESRD, which was almost certainly the first recognition of hyperlipidemia.13 Later, the dyslipidemia of CRI was described by Baghdad and Samkelson, who reported that hypertriglyceridemia was a characteristic of patients on hemodialysis.6
CRI produces characteristic effects on major lipoprotein fractions, which can be summarized as increased VLDL, chylomicrons and IDL remnants1,8,12 with high triglyceride concentrations, low HDL cholesterol concentrations,1,4,6,8,13,14 and with total cholesterol and LDL-cholesterol concentrations 1,6,8,13,14 near normal or even slightly lower than those in the general population.1,13,14
Triglyceride elevation is present in up to 70% of patients with ESRD, but hemodialysis tends to improve triglyceridemia, at least in nondiabetic patients.13 In contrast, patients on peritoneal dialysis tend to exhibit hypercholesterolemia when compared with patients on pre-dialysis or those on hemodialysis, due to the usual increase in total cholesterol and LDL-cholesterol.1 Therefore, although the authors have chosen to focus on dyslipidemia in CRI in general, there may be some differences in the lipid profile of patients undergoing hemodialysis, peritoneal dialysis, and in those with nephrotic syndrome (Table 1).4
Lipid profile in patients with chronic renal insufficiency undergoing dialysis or with nephrotic syndrome.1,4
| CRI | Nephrotic syndrome | Hemodialysis | Peritoneal dialysis | |
| Total cholesterol | ↔ | ↑↑ | ↔, ↓ | ↑, ↔ |
| LDL cholesterol | ↔ | ↑↑ | ↔, ↓ | ↑, ↔ |
| HDL cholesterol | ↓ | ↓ | ↓ | ↓ |
| Non-HDL cholesterol | ↑ | ↑↑ | ↔, ↓, ↑ | ↑ |
| Triglycerides | ↑ | ↑↑ | ↑ | ↑ |
| Lp(a) | ↑ | ↑↑ | ↑ | ↑↑ |
Normal (↔), increased (↑), markedly increased (↑↑), and decreased (↓) plasma levels compared with non-uremic individuals.
LDL: low density lipoproteins; HDL: high density lipoproteins; Lp(a): lipoprotein (a); CRI: chronic renal insufficiency.
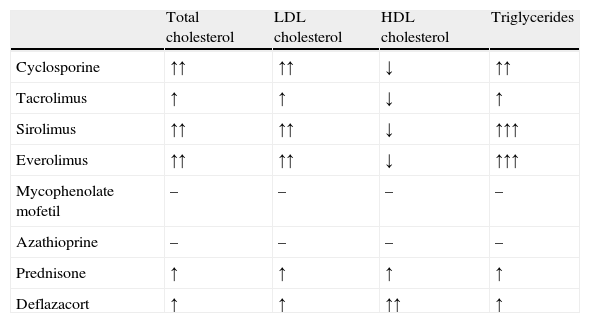
Before we review the pathophysiology of dyslipidemia in CRI, dyslipidemia following kidney transplantation should be briefly discussed. Indeed, lipid abnormalities are a common complication of kidney transplantation, occurring in 20–63% of posttransplant patients.15 Impairment of lipid metabolism is often present before renal transplantation. After transplantation and renal function recovery, lipid disturbances usually persist but show a different profile due to the various effects of immunosuppressive drugs,16,17 such as calcineurin inhibitors (cyclosporine and tacrolimus), antiproliferative drugs (mycophenolate mofetil and azathioprine), mammalian target of rapamycin inhibitors (sirolimus and everolimus), and corticosteroids.16 The most frequent alterations in the lipid profile of renal transplant recipients are elevated total cholesterol, LDL cholesterol and triglyceride concentrations, and decreased HDL cholesterol concentrations,15 although an increase in HDL cholesterol is frequently observed in patients treated with corticosteroids, such as prednisone and deflazacort (Table 2).16
Effect of immunosuppressive drugs on lipid parameters.16
| Total cholesterol | LDL cholesterol | HDL cholesterol | Triglycerides | |
| Cyclosporine | ↑↑ | ↑↑ | ↓ | ↑↑ |
| Tacrolimus | ↑ | ↑ | ↓ | ↑ |
| Sirolimus | ↑↑ | ↑↑ | ↓ | ↑↑↑ |
| Everolimus | ↑↑ | ↑↑ | ↓ | ↑↑↑ |
| Mycophenolate mofetil | – | – | – | – |
| Azathioprine | – | – | – | – |
| Prednisone | ↑ | ↑ | ↑ | ↑ |
| Deflazacort | ↑ | ↑ | ↑↑ | ↑ |
Normal (↔), increased (↑), markedly increased (↑↑), and decreased (↓) plasma levels.
LDL: low density lipoproteins; HDL: high density lipoproteins.
The general mechanisms underlying the dyslipidemia of renal disease may be summarized as altered metabolism of postprandial lipoproteins, altered metabolism of other triglyceride-rich lipoproteins (TRL), changes in the route of reverse cholesterol transport, structural changes of lipoproteins, postribosomal modifications of lipoproteins, insulin resistance, proteinuria, and increased lipoprotein(a).
Altered metabolism of postprandial lipoproteinsIn CRI there is an increase in postprandial triglycerides and abnormal prolongation of this increase. The disturbance of the clearance of chylomicron remnants may underlie this change.1,12 However, since chylomicrons compete with TRL of hepatic origin for lipolysis by LPL, the postprandial increase in triglycerides in the circulation is worsened by TRL of hepatic origin. Thus, the postprandial increase in triglycerides involves not only the metabolism of chylomicrons of intestinal origin but also the metabolism of TRL of hepatic origin.1
Altered metabolism of other triglyceride-rich lipoproteinsThe main mechanism of TRL increase in CRI appears to be a decrease in lipolysis,1 due to reduced activity of major vascular endothelium associated lipases, LPL and hepatic lipase.1,11,13 This reduced activity leads to the accumulation of their remnants (chylomicron remnants, VLDL remnants, IDL)4,13 containing apo-B and also apo-C and apo-E in comparison with lipoproteins containing apo-A.6 The decrease in lipolytic activity may result from increased concentrations of different inhibitors in circulation,4 including apo-CIII,1,6 which can modulate the binding of lipoproteins to cell receptors, and is a potent inhibitor of LPL1,6 and immature HDL (composed of apo-AI) not found in the lipoprotein fraction of normal plasma. The disruption of lipase activity may also be caused by an inhibitory effect of hyperparathyroidism associated with renal disease, which leads to an accumulation of calcium in pancreatic islet cells and subsequent reduction in insulin secretion, or may derive from the depleted pool of enzymes by frequent heparinization during hemodialysis.11 In addition, lipogenesis,13 namely VLDL synthesis,1 also seems to be slightly increased.
HDL and changes in the route of reverse cholesterol transportIn patients with CRI, HDL and apo-AI concentrations are almost universally decreased.1,4,8,18 Indeed, the increase in TRL with subsequent transference of triglyceride excess to HDL increases its susceptibility as a substrate to hepatic lipase,1,8 leading to a decrease in its size, originating small and dense HDL.8 In addition, these lipoproteins spill apo-AI, which is lost by glomerular filtration and renal catabolism.1
Other mechanisms may contribute to the low HDL concentrations in CRI, such as decreased activity of lecithin-cholesterol acyltransferase,1,4,8,13,18 which is associated with delayed maturation of HDL,8 since this enzyme normally increases the uptake of esterified cholesterol by this lipoprotein.13
The enzyme paraoxonase, carried by HDL, is also reduced among patients with CRI,4,8,18 although the reason is unknown. Consequently, HDL in patients on dialysis has low antioxidant activity.8
Notably, the structural changes that lead to the reduced capacity of HDL to transport cholesterol disturb the reverse transport of lipids from the periphery to the liver.4,11
Structural changes of lipoproteinsThe incomplete catabolism of lipoproteins, mentioned above, leads to the accumulation of lipoprotein remnants, which contribute to the heterogeneity of TRL present in plasma, with different backgrounds, sizes, compositions, and degrees of atherogenicity.4 Hypertriglyceridemia associated with CRI is also expressed in terms of individual classes of lipoproteins––the ratio of triglycerides to esterified cholesterol is higher in LDL and HDL and lower in VLDL and IDL.1 These changes are consistent with a scenario common to all states of hypertriglyceridemia––a pathological increase of TRL is followed by the transference of triglycerides from the expanded pool of substrate to HDL and LDL in exchange for esterified cholesterol by the action of enzyme cholesteryl ester transfer protein.1,8 Thus, despite normal or lower LDL cholesterol, this lipoprotein tends to be smaller and denser than normal,1,8,13 due to increased VLDL precursors and the exchange of triglycerides for esterified cholesterol followed by triglyceride lipolysis.1
In the context of uremic microinflammation,13 HDL also undergoes structural changes through the incorporation of serum amyloid A, resulting in the so-called acute phase or inflammatory HDL,13,14,18 which act not as protective particles, but as pro-atherogenic particles.4,12,14
Postribosomal modifications of lipoproteinsThe increase in residence time of lipoproteins in the circulation, due to their decreased catabolism, predisposes them to postribosomal modifications,7,11 such as oxidation, glycation, and carbamilation4,6,18 with a consequent reduction in the recognition and binding to their receptors4,12,13 and increased atherogenicity.6 However, in contrast with the decreased hepatic clearance, there is increased clearance by the scavenger pathway. The modified LDL, for example oxidized, are captured by macrophages by scavenger receptors, which may lead to the transformation of these cells into foam cells, which are associated with atherogenesis.4
The plasma activity of paraoxonase is also reduced in CRI,4,18 predisposing LDL,4,12 and possibly HDL, to oxidation.4 Furthermore, since small and dense LDL are prone to oxidation, and as oxidative stress is increased in CRI, oxidized LDL concentrations are also increased.1
Insulin resistanceInsulin resistance develops early in renal insufficiency6 and is a frequent feature in CRI, most likely secondary to a postreceptor insulin defect. Additionally, many individuals with uremia show a decreased insulin secretory response1 and insulin resistance per se is associated with decreased GFR.8 The pattern of dyslipidemia found in CRI is similar to that associated with insulin resistance.1 Indeed both insulin resistance and CRI are associated with low HDL and increased triglycerides.8
Importantly, insulin has a regulatory role in LPL and therefore changes in insulin secretion or sensitivity may play a significant role in the development of dyslipidemia in the context of uremia.1 Moreover, with regard to free fatty acid efflux from adipose tissue, there are two major intracellular lipases: adipocyte triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL). Although the regulation of ATGL is not yet well defined, inhibition of HSL by insulin is a well known phenomenon that plays a critical role in energy balance. Thus, insulin resistance is associated with disturbed inhibition of diacylglycerol hydrolysis by HSL, eventually leading to excess release of free fatty acids from adipocytes.1
Another possible link between insulin resistance and dyslipidemia in CRI may be the decreased alpha-type peroxisome proliferator-activated nuclear receptor (PPARα) activity, which is associated with increased triglycerides and VLDL concentrations. In addition, decreased expression of the PPARα gene may result in decreased LPL activity and increased production of apo-CIII with a consequent delay in the catabolism of TRLs.6
ProteinuriaHyperlipidemia of the nephrotic syndrome is clinically obvious. There is growing evidence that non-nephrotic proteinuria also affects the physiology of lipoproteins. Several studies have shown an association between microalbuminuria and dyslipidemia, as well as with other components of metabolic syndrome.1 In fact, patients with diabetes mellitus or hypertension with macroalbuminuria or microalbuminuria have a higher rate of dyslipidemia than those with normoalbuminuria. Since CRI is often associated with non-nephrotic proteinuria, this could be a mediator of uremic dyslipidemia.1
Increase of lipoprotein (a)Lp(a) is a lipoprotein similar to LDL in lipid and protein composition, but additionally contains apo(a), which is synthesized in the liver and is linked to apo-B-100 by a disulfide bridge.2–5,8 This lipoprotein has high homology with plasminogen,2,4,5 interfering with fibrinolysis, and also binds to macrophages, promoting the formation of foam cells.11
Plasma concentrations of Lp(a) are strongly genetically determined by the apo(a) gene. Individuals with apo(a) isoforms of high molecular weight usually have low mean Lp(a) concentrations, whereas those with isoforms of low molecular weight usually exhibit more elevated plasma concentrations of Lp(a).4
In addition to the genetic influence, Lp(a) concentrations are also affected by the GFR in renal disease.4 These concentrations are increased in CRI8,13 and in nephrotic syndrome. In the first case, this increase appears to result from decreased catabolism,8,14 while in the second, it results from increased synthesis.8
The type of dialysis may also be important––concentrations are higher in patients treated with continuous ambulatory peritoneal dialysis than in those under hemodialysis. This effect may be mediated, at least in part, by an increased production of cytokines in the first type of dialysis. In contrast, successful kidney transplantation is usually associated with a reduction in Lp(a) levels.19
Lp(a) is a potent cardiovascular risk factor in the general population4,8,11–13,18 and the risk is associated with its concentration and small size.8,12
ConsequencesCardiovascular diseaseCRI is increasingly being noted to be an independent risk factor for the development of coronary artery disease. The development of accelerated atherosclerosis and cardiovascular disease (CVD) in progressive renal disease is well documented.6 CVD is the leading cause of mortality during dialysis and after renal transplantation. The risk of cardiovascular mortality in ESRD is about 10 times higher than in the general population.20 The prevalence of CVD is also high in patients in the early stages of CRI,6,13 long before they start on dialysis.5
However, the association of dyslipidemia with cardiovascular events in patients with end-stage renal disease (ESRD) has not been uniformly observed. One of the most powerful confounding variables is microinflammation.13 The risk of cardiovascular events is high in ESRD,20,21 but whether this is due to lipids or is due to other cardiovascular risk factors, such as fibrinogen, Lp(a), advanced glycosylation end products, and phosphate, is unclear.21 Indeed, there are several factors that could contribute to the risk of CVD associated with dyslipidemia in CRI. The atherogenic potential of dyslipidemia in CRI may be more dependent on apoprotein than lipid abnormalities and may not always be identified by measurement of plasma lipids alone.4 The proinflammatory effects of apo-CIII on monocytes and endothelial cells, for example, appear to play a critical role in the development of atherosclerosis in CRI.6 This apolipoprotein alone or in lipoproteins containing apo-B stimulates the peripheral adhesion of monocytes to endothelial cells and also induces the expression of adhesion molecules on endothelial cells, inhibits insulin signaling in the endothelium, and attenuates insulin-stimulated endothelial nitric oxide synthase, thereby impairing the endothelium relaxation normally induced by nitric oxide.6 Abundant vascular calcifications have been described in uremic patients9,12,22 and cardiovascular risk factors (high fibrinogen, Lp(a), homocysteine, glucose, oxidative processes) may appear early in the course of renal disease.12
In the MARS study (Monitored Atherosclerosis Regression Study), elevated levels of apo-B-containing and apo-C-containing TRL, in general, and Lp-A-II:B:C:D:E particles, in particular, significantly contributed to the progression of coronary artery disease.23 In turn, the CARE (Cholesterol and Recurrent Events) study showed that an increased concentration of apo-CIII in VLDL and IDL was a strong predictor of coronary events.24
Progression of renal diseaseCRI tends to progress but the rate at which this occurs may vary depending on factors such as the presence of concomitant disease (for example diabetes mellitus) or glomerular proteinuria.25 Even healthy individuals show a decline of renal function over time. Above 30 years, the average GFR reduction is about 0.7 to 0.9mL/min/year. Dyslipidemia may perpetuate or accelerate this decline.26
The deposition of lipids in the kidney may cause kidney damage in a manner analogous to lipid deposition in the vascular wall in atherosclerosis development.12,25
In 1982, Moorhead et al were the first to hypothesize that hyperlipidemia impairs renal function. Since then new evidence has come to light that supports this hypothesis, due to damage of mesangial cells, endothelial cells and glomerular podocytes. LDL can activate receptors expressed by mesangial cells, stimulate the production of matrix proteins and promote the generation of proinflammatory cytokines, which can lead to the recruitment and activation of macrophages. The accumulation of lipoproteins in glomerular mesangium may also promote matrix production and glomerulosclerosis. Podocytes can be damaged by triglycerides and cholesterol. Oxidized LDL can stimulate monocyte infiltration.10 The capture of oxidized lipoproteins via scavenger receptors, the reabsorption of lipids binding filtered proteins via megalin–cubilin complex, and the increased uptake of glucose by the nephron may promote the glomerular, tubular, and interstitial accumulation of lipids.27 In addition, the continued loss of functioning nephrons, with subsequent hypertension and hyperfiltration in the remaining nephrons, may worsen renal function.6
TreatmentTheoretically, initiation of therapy aimed at controlling CRI-associated dyslipidemia may help to mitigate its possible renal and cardiovascular consequences.6 However evidence of benefit is still lacking.1 Moreover, the best treatment option in CRI has not yet been well defined.11,12 In the general population, clinical trials have shown that cardiovascular mortality decreased in proportion to the rate of reduction of LDL cholesterol. However, there are few data on the contribution of dyslipidemia to cardiovascular morbidity and mortality in CRI.11 Some authors argue that the dyslipidemia of CRI should be treated as a coronary heart disease risk equivalent similar to diabetes mellitus and that the cut-off for the decision to treat should be decreased given the high risk that it entails.9,14
In turn, the National Kidney Foundation (NKF)/Kidney Disease Outcomes Quality Initiative guidelines on the management of dyslipidemia in CRI recommend that treatment should be considered in patients with CRI stage 5 and LDL cholesterol >100mg/dL to reduce LDL cholesterol to less than 100mg/dL. Treatment should also be considered in individuals with LDL cholesterol<100mg/dL but triglyceride concentrations >200mg/dL and non-HDL cholesterol >130mg/dL.22 Although patients with ESRD have high rates of cardiovascular disease, the demanding LDL targets set by the NCEP (National Cholesterol Education Program) ATPIII (Adult Treatment Panel) 2004 have not yet been adopted for patients with ESRD. Indeed, to achieve these objectives most patients will need drugs.22
StatinsStatins inhibit 3-hydroxy-3-methylglutaryl coenzyme A reductase11,22 and are the most widely studied drug class in the treatment of dyslipidemia in ESRD.22 In the general population, these drugs have shown clear benefits in cardiovascular prognosis, both in primary prevention studies and in secondary prevention.10,11,25 However, there is no conclusive evidence that statins improve cardiovascular prognosis in patients with CRI.11,25 As in the general population, statins are highly effective in lowering total cholesterol and LDL in uremic patients,4,11 but their effectiveness in reducing cardiovascular events may vary depending on the stage of CRI.4
Moreover, statins are generally ineffective in correcting hypertriglyceridemia, Lp(a) elevation and the decrease in HDL, which are the main changes in the dyslipidemia of CRI.4,6 Therefore, nicotinic acid and, to a lesser extent, fibrates – at least in theory – may be the best therapeutic options in uremic dyslipidemia. However, there are no studies of the effectiveness of these agents in improving cardiovascular disease in patients with CRI.4
Most studies examining the effect of lipid reduction have excluded patients with CRI.1 Analysis of large prevention trials using statins has allowed limited definition of subsets of patients with CRI1,4,26 but the results are contradictory.1,6 Some studies have shown a cardiovascular benefit of this treatment, such as the TNT (Treatment to New Targets),28 HPS (Heart Protection Study),29 and a combined analysis of the CARE (Cholesterol And Recurrent Events), WOSCOPS (West Of Scotland Coronary Prevention Study), and LIPID (Long-term Intervention with Pravastatin in Ischemic Disease) studies.30 On the other hand, the ALLHAT study (Antihypertensive and Lipid Lowering treatment to prevent Heart Attack Trial) found no beneficial effect of lipid-lowering therapy.31 Isbel et al,32 who conducted a randomized controlled study in 200 patients with advanced CRI comparing standard therapy with careful treatment of different cardiovascular risk factors, including treatment with statins, found no differences in outcome after 2 years.
Three randomized controlled trials on statin therapy were completed in patients with renal function replacement treatment (hemodialysis or renal transplantation). There are no controlled intervention studies in patients on peritoneal dialysis. Both the 4D (Die Deutsche Diabetes-Dialyse) study in 1255 patients with type 2 diabetes on dialysis,33 or the AURORA (A study to evaluate the Use of Rosuvastatin in subjects On Regular hemodialysis; an Assessment of survival and cardiovascular events) study in 2776 patients on dialysis due to different etiologies,34 showed that statins had no effect on mortality from cardiovascular disease or on all-cause mortality.4,6 The ALERT (Assessment of Lescol in Renal Transplantation) study on the use of fluvastatin in kidney transplant recipients was also unable to show significant effects on the primary outcome of cardiovascular disease.35 However, the analysis of a subgroup of patients in this study suggested a reduction in the incidence of myocardial infarction and further follow-up of this study reported a significant reduction in major cardiovascular events after an average follow-up of 6.7 years.6 Notably, for some authors, the results from the 4D and Aurora studies should not be extrapolated to patients with mild to moderate CRI.26
Currently, there are two ongoing studies to evaluate the effect of decreasing lipids in patients with CRI by using statins. The SHARP (Study of Heart And Renal Protection)36 trial includes dialysis patients and patients with CRI in earlier stages, randomized to treatment with simvastatin 20mg in combination with ezetimibe 10mg/day or to placebo,1,6,22 and the results are expected in 2010.6 This study involves the recruitment of 6000 patients with CRI with serum creatinine >1.7mg/dL in men and >1.5mg/dL in women and 3000 patients undergoing dialysis.11 More recently, the smaller LORD (Lipid lowering and Onset of Renal Disease) study37 was initiated in Australia and includes randomization of patients with CRI to atorvastatin or placebo and is planned to last 3 years. The SHARP study has cardiovascular disease morbidity as the primary outcome variable, whereas the primary aim of the LORD trial is to assess the effect of statins on the progression of kidney disease.6
With regard to the benefit of statins on the kidney, some studies suggest that these drugs slow the rate of renal function decline in patients with mild to moderate impairment, while others show no more effect than placebo.26 A positive effect of statins on renal disease progression was reported in the CARE (Cholesterol And Recurrent Events) study, which recruited 4159 hyperlipidemic individuals with a history of acute myocardial infarction, who were randomly treated with pravastatin or placebo and their cardiovascular morbidity and mortality was followed for 5 years. A retrospective analysis of a subgroup of 690 patients with GFR<60mL/min showed that the loss of GFR in the pravastatin group did not differ from that in the placebo group, but statins were beneficial in those who had proteinuria at baseline and those with lower basal GFR. The possible benefit was associated with improvement in inflammation markers. In addition, analysis of a subgroup of patients in the TNT study, which compared the effect of 80 versus 10mg of atorvastatin, after a follow-up of 5 years showed an increase in GFR in both groups and this increase was higher in the 80mg group (5.2 versus 3.5mL/min).26
The renoprotective effect of statins may not be only due to lipid reduction, but also to a decrease of renal interstitial inflammation,10,11,22,26 improvement of renal hemodynamics, and a decrease in glomerular proteinuria.10,11 Statins may also have a beneficial effect on vascular stiffness and endothelial function in renal failure.26
However, other studies have shown that statins do not reduce renal dysfunction progression: a secondary analysis of the PREVEND-IT (Prevention of Renal and Vascular End-stage Disease Interventional Trial)38 study, which included 864 patients with albuminuria and relatively preserved GFR, showed no change in GFR between pravastatin and placebo in 4 years, despite a reduction of cholesterol in the first group. A retrospective analysis of the ALLHAT31 study showed that pravastatin and placebo had similar effects on the rate of ESRD and GFR reduction over a period of 6 years.26,31 Many authors argue that statins should be administered to all patients with CRI and ESRD because of their high safety profile and beneficial effect even after kidney transplantation, although this benefit has not yet been demonstrated in prospective studies.13 However, other researchers warn of the risk of increased adverse effects with the use of statins in CRI, for example liver damage and rhabdomyolysis.4,11,14
Current recommendations are to reduce the statin dose by 50% in dialysis patients, except for atorvastatin and pravastatin.11,22 In transplant recipients, a lower starting dose of statins should be used, particularly with concomitant use of cyclosporine.11
FibratesFibrates are PPARα activators.1,6,22 Activation of this transcriptional factor increases oxidation of fatty acids in the liver, kidney, heart, and skeletal muscle and reduces hepatic production of apo-CIII and increases expression of LPL,22 apo-AI, and apo-AII,1 as well as increases HDL and lowers total cholesterol, triglycerides, and LDL.22 Given the early development of hypertriglyceridemia in CRI, fibrates would be logical candidates for the treatment of dyslipidemia in this disease, but are not often used because of the risk of rhabdomyolysis,13 worsening of renal function,1,22 impairment of liver function, and elevation of homocysteine in these patients.1
The NKF guidelines favor the use of gemfibrozil because this drug does not requires dose adjustment for the decrease in GFR and its pharmacokinetics are not altered in this context.1 These guidelines discourage the use of fibrates in patients with GFR<15mL/min.11,22 Several observational studies suggest that statins, and even fibrates, by reducing LDL, triglycerides, or both, may reduce the relative risk of coronary heart disease and general mortality from CVD.6 In the FIELD study (Fenofibrate Intervention and Event Lowering in Diabetes), fibrates reduced overall CVD in diabetics, but their effect on coronary events was disappointing.39 Some studies with gemfibrozil have associated this drug with a 20% reduction in cardiovascular events in patients with clearances between 30 and 75mL/min (mild–moderate CRI).4,11 However, randomized controlled trials on the potential effectiveness of fibrates in patients with CRI are still lacking.4
Nicotinic acidNicotinic acid is highly effective in increasing HDL4 by decreasing cholesterol transfer from HDL to VLDL and also by decreasing HDL clearance22 and is the only available drug to substantially lower plasma Lp(a).4 This drug also reduces VLDL secretion by the liver.11
There are two formulations of nicotinic acid on the market, a sustained-release form and an immediate-release form.19 The immediate-release formulations caused flushing, itching, rash, nausea, and gastrointestinal adverse effects, but these effects are less frequent with the new sustained release formulations.4,22 Compliance can be increased by concurrent administration of aspirin and gradual dose increment.4
In the general population nicotinic acid has been shown to improve cardiac and cerebrovascular prognosis. Studies in CRI are small and of short duration, but showed the expected changes in lipid profile. No studies have examined their impact on cardiovascular prognosis in patients with CRI.4 The use of this drug in dialysis patients was investigated mainly in Japan. Although there are studies in which there were no serious adverse effects, statins were superior in reducing LDL.22
Nicotinic acid can be considered an alternative second line agent in patients who cannot tolerate statins.22 Indeed, the NKF guidelines recommend nicotinic acid as a second line agent for reducing LDL in combination with a statin.11
Bile acid binding resinsThese drugs bind to bile acids in the intestine and reduce their enterohepatic circulation, with a subsequent increase in the conversion of cholesterol to bile acids in the liver, an effect of feedback regulation. This results in increased expression of LDL receptors in the liver with a resultant increase of LDL particle clearance. These drugs can reduce LDL cholesterol by 10–20% in the general population, but in some individuals may increase hepatic production of VLDL,22 with a consequent increase in plasma triglycerides.11,22 Because bile acid binding resins are not absorbed in the gastrointestinal tract, the dose does not need to be adjusted in patients with decreased kidney function;11,22 these drugs do, however, interfere with the absorption of cyclosporine (which may be an important issue in transplant recipients).11
Bile acid binding resins have already demonstrated a 19% reduction in cardiovascular mortality in asymptomatic individuals with normal renal function. There are few data on their safety and prognosis in CRI.11 Reported adverse effects are gastrointestinal, mainly constipation, and interference with drugs such as digoxin and warfarin.22
SevelamerSevelamer is a phosphate binder that also effectively binds bile acids, allowing plasma cholesterol to be lowered.1,11,22 This agent can reduce total cholesterol and LDL by 18–22% and 30–37%, respectively.11 Treatment with this drug is also associated with a reduction in the concentration of phosphorus and calcium phosphorus product,22 but it is unclear whether this results in reduced progression of coronary calcification.4 There are no long-term studies on the cardiovascular benefits of its use.11
EzetimibeEzetimibe has low interaction with other drugs and can therefore be used in conjunction with statins or other lipid-lowering drugs. Coadministration of cholestyramine decreases its bioavailability, while the concomitant use of gemfibrozil and fenofibrate slightly increases its availability. This agent can reduce cyclosporine levels.22
The combination of ezetimibe 10mg/day with a statin also produces a greater reduction in C-reactive protein concentrations than a statin alone.22
Other drugsThe omega-3 fatty acids eicosapentaenoic and docosahexaenoic found in fish oils reduce VLDL production and are used therapeutically in patients with hypertriglyceridemia. These fatty acids could therefore be expected to improve dyslipidemia in CRI, although some studies showed no benefit over placebo. Other studies showed that in patients on dialysis, these acids can improve the lipid profile in most – but not in all – patients.1
Finally, antioxidants such as vitamin E may have a beneficial effect on the susceptibility of LDL to oxidation in patients on dialysis. Some studies suggest that vitamin E may increase HDL in patients on dialysis.11,22 Randomized studies in the general population which used antioxidants such as vitamin E showed no cardiovascular benefit.11 However, in an assay with 196 hemodialysis patients with cardiovascular disease, Boaz et al40 found that vitamin E given for an average of 519 days reduced final mortality due to myocardial infarction, peripheral vascular disease, and ischemic stroke by 54% when compared with placebo. Randomized studies are needed to confirm these results.11
DietThe general nutritional guidelines for dyslipidemic patients recommend reducing intake of total, saturated, and trans fat, as well as limiting consumption of cholesterol, which should be coordinated with the dietary guidelines in patients with CRI.1 Thus, dietary counseling should be cautious and given with the support of an experienced nutritionist due to the high prevalence of malnutrition in ESRD. There are no prospective clinical studies that have investigated the long-term benefit of lifestyle intervention in ESRD.22
ConclusionsDyslipidemia in CRI is mainly characterized by increased triglycerides and reduced HDL with almost normal levels of total and LDL cholesterol. However, lipoprotein concentrations may not adequately reflect the dyslipidemia, because in addition to changes in lipoprotein levels, there are also alterations in their composition and function. The most important pathophysiologic factors in this process seem to be altered metabolism of lipoproteins, postribosomal modifications, insulin resistance, non-nephrotic proteinuria, and increased apo-CIII.
There is some evidence that dyslipidemia in CRI is a risk factor for cardiovascular and renal disease progression, but there are no conclusive studies on optimal lipid goals and no clear statements on the most effective treatment option for this disorder.
However, ongoing studies are likely to help clarify the impact of dyslipidemia in renal disease and the benefits of establishing appropriate lipid-lowering therapy.
Conflict of interestThe authors have no conflicts of interest.
