


Los craneofaringiomas son tumores benignos poco frecuentes originados a partir de restos embrionarios de la bolsa de Rathke. La edad de presentación sigue una distribución bimodal (5–14 años y 50–74 años)1. Su localización más habitual es la región selar y paraselar. Clínicamente se caracterizan por síntomas derivados de la compresión local de estructuras adyacentes asociados o no a hipopituitarismo parcial o total. Su tratamiento de elección es el quirúrgico, siendo poco habitual la recuperación hormonal tras la cirugía1. La enfermedad de Graves es la causa más frecuente de hipertiroidismo, con un pico de incidencia entre los 40–60 años2. Su asociación con patología tumoral hipofisaria no es habitual. Presentamos un nuevo caso de hipertiroidismo por enfermedad de Graves desarrollado 6 meses después del tratamiento quirúrgico de un craneofaringioma.
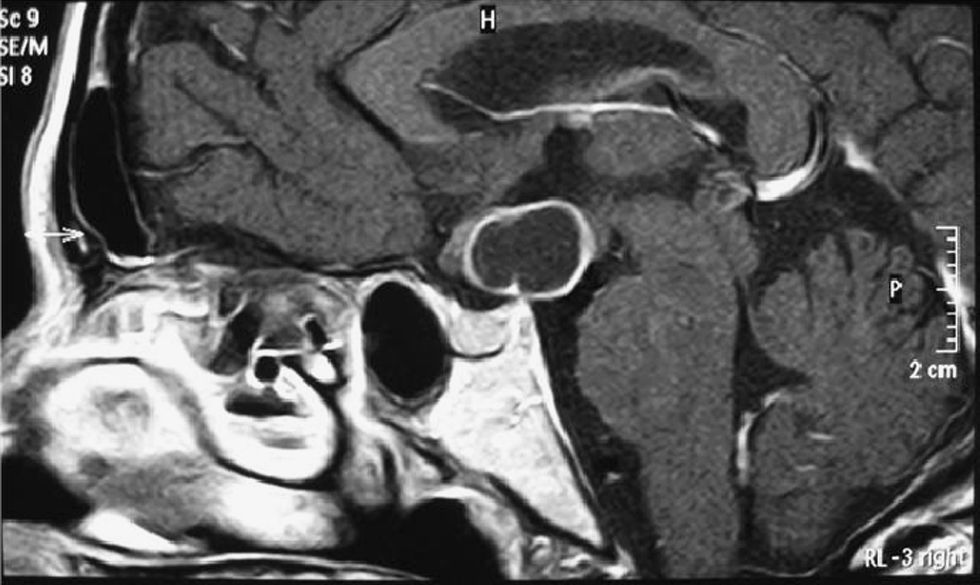
Mujer de 49 años, sin antecedentes de interés, que acude a urgencias por cefalea y pérdida de campo visual en territorio temporal izquierdo de 6 meses de evolución. El estudio morfológico mediante tomografía axial computarizada (TAC) craneal mostró una lesión supraselar hipodensa de 2,0×1,8cm. La resonancia magnética (RM) cerebral reveló una lesión quística con engrosamiento de quiasma óptico y obliteración del tercer ventrículo sugerente de craneofaringioma (fig. 1). La campimetria confirmó una hemianopsia heterónima bitemporal. El estudio hormonal puso de manifiesto una hiperprolactinemia (prolactina[PRL]: 130ng/ml; valores normales [N]: 3–24ng/ml) con hipogonadismo hipogonadotropo (folitropina [FSH]: 1,42mUI/ml, N en menopausia: 26,7–133,4mUI/ml; lutropina [LH]:<0,07mUI/ml, N en menopausia: 10,4–64,5mUI/ml) y estradiol <10pg/ml, N en menopausia: <10–28pg/ml), insuficiencia suprarrenal (cortisol basal <1μg/dl, N: 5–25mcg/dl) y probable déficit de somatotropina (GH): factor de crecimiento insulínico tipo 1 (IGF-1): 80,7ng/ml, límites de referencia por edad: mediana 154ng/ml (rango: 94-252ng/ml). Tan solo el eje hipotálamo-hipófiso-tiroideo se encontraba conservado (tirotropina [TSH]: 0,370μU/ml, N: 0,350–4,959mcU/ml; tiroxina libre [T4L]: 0,83ng/dl, N: 0,70–1,48ng/dl y triyodotironina libre [T3L]: 1,71pg/ml; N: 1,71–4,53pg/ml). La paciente fue intervenida mediante craniectomía pterional derecha. La reevaluación hormonal postquirúrgica fue compatible con hipopituitarismo parcial: déficit de TSH (TSH 0,017μU/ml, T4L: 1.01ng/dl y T3L 3,94pg/ml), FSH/LH (LH<0,07mUI/ml, FSH 1,14mUI/ml y estradiol<10pg/ml), GH (GH basal 0,59ng/ml e IGF-1 67ng/ml) y corticotropina (ACTH) (test de Nuvacthen: cortisol basal 1,18mcg/dl que asciende a 11,2mcg/dl a los 60min con ACTH 6,53pg/ml), hiperprolactinemia (PRL: 136ng/ml) y diabetes insípida central completa (osmolalidad sanguínea de 304mOsm/Kg y urinaria de 58mOsm/kg a las 6h de la prueba de deshidratación, y osmolalidad urinaria de 268mOsm/kg tras 2mcg sc de desmopresina). La paciente inició tratamiento sustitutivo con glucocorticoides (hidroaltesona 20-0-10mg), levotiroxina (50μg/24h) y desmopresina (0,1mg/8h, vo). Seis meses después del alta acudió al servicio de urgencias por astenia intensa y un cuadro presincopal. En la exploración destacaba la presencia de taquicardia (frecuencia cardiaca 106lpm), piel húmeda y caliente y retracción palpebral bilateral con exoftalmos y quemosis del ojo izquierdo. Se palpaba un bocio difuso grado 2 con thrill. La analítica demostró hipertiroidismo (TSH suprimida, T4 libre 3,65ng/dl y T3 libre >30pg/ml), confirmada tras retirar la levotiroxina. El estudio inmunológico fue negativo para anticuerpos antiperoxidasa tiroidea y antitiroglobulina y positivo para anticuerpos frente al receptor de TSH (TSI) 7,56U/l (positivos >1,5U/l). La gammagrafía mostró un tiroides aumentado de tamaño con hipercaptación global. Se inició tratamiento con antitiroideos (metimazol 30mg/d, vo) y se aumentó la hidroaltesona a dosis de estrés hasta normalización de la función tiroidea, momento en el que se instauró tratamiento definitivo con radioyodo.

El presente caso clínico nos muestra una mujer de 49 años portadora de un craneofaringioma e hipopituitarismo parcial compresivo que desarrolla un hipertiroidismo por enfermedad de Graves a los 6 meses de la intervención quirúrgica del tumor.
La concurrencia de hipertiroidismo y patología hipotálamo hipofisaria con hipopituitarismo no es habitual. El hipertiroidismo se ha descrito asociado a hipopituitarismo de diferentes causas como hipofisectomía quirúrgica3, sección del tallo hipofisario por exoftalmos maligno4, necrosis hipofisaria postparto (síndrome de Sheehan)5, destrucción hipofisaria por adenoma cromófobo6 y radioterapia de tumor nasofaríngeo con invasión hipofisaria7 desarrollando posteriormente un hipertiroidismo. En algunos de ellos el desarrollo del hipertiroidismo apareció 8 años después7.
Hasta la fecha la aparición de hipertiroidismo e hipopituitarismo por craneofaringioma se ha descrito de forma excepcional8–11 y, tan sólo en 3 casos, se ha asociado claramente a hipertiroidismo por enfermedad de Graves8–10. El primero de ellos fue descrito en 1964 por Taunton y Pittman8, se trataba de un varón de 20 años que debutó con cefaleas y clínica de hipogonadismo y hemianopsia homónima derecha. Fue diagnosticado de tumoración supraselar e hipogonadismo hipogonadotropo. Tras la intervención persistió la deficiencia de gonadotropinas y desarrolló un hipotiroidismo central iniciando tratamiento hormonal sustitutivo. Un año y medio más tarde desarrolló un hipertiroidismo con bocio difuso hipercaptante que fue controlado con antitiroideos. El segundo caso9 fue comunicado en nuestro país en 2008; se trataba de una mujer de 88 años que debutó con un panhipopituitarismo e hiperprolactinemia desarrollando un hipertiroidismo por enfermedad de Graves a partir del hipotiroidismo central. El estudio morfológico mediante RM y TAC mostró un proceso expansivo de localización selar y supraselar compatible con craniofaringioma. La paciente inició tratamiento sustitutivo y antitiroideos durante 9 meses con remisión de la enfermedad de Graves. El último caso también descrito en 200810 era un varón de 46 años intervenido de un craniofaringioma con hipopituitarismo persistente tras cirugía que presentó una crisis suprarrenal aguda como consecuencia del desarrollo de un hipertiroidismo por enfermedad de Graves. El paciente fue tratado con tratamiento hormonal sustitutivo y antitiroideos hasta la normalización de la función tiroidea y luego fue remitido a dosis terapéutica de radioyodo. Es posible que, en los casos descritos de aparición de enfermedad de Graves tras la cirugía del craneofaringioma, la intervención quirúrgica del tumor constituyó el factor de estrés desencadenante para el inicio del hipertiroidismo autoinmune en pacientes genéticamente susceptibles12.
En el momento actual se desconoce si existe algún vínculo entre la enfermedad de Graves y el craneofaringioma y/o el hipopituitarismo asociado como factores precipitantes del hipertiroidismo autoinmune.
El manejo clínico de ambas entidades sería el mismo que cuando aparecen por separado, con la precaución de evitar el tratamiento con levotiroxina y realizar cobertura esteroidea en períodos activos del hipertiroidismo por enfermedad de Graves. Parece razonable que en estos casos el tratamiento definitivo del hipertiroidismo mediante radioyodo o cirugía sea la opción terapéutica de elección con la finalidad de facilitar el manejo del hipopituitarismo y evitar complicaciones como crisis suprarrenales o descompensación de la diabetes insípida en los casos de recidiva del hipertiroidismo.
