



Presentamos un paciente con enfermedad de Behçet de larga evolución, con predominio de manifestaciones neurológicas, que ingresó por insuficiencia suprarrenal, en el que se demostró déficit aislado de corticotropina (DAACTH). El DAACTH es una característica típica de las hipofisitis y se ha descrito en asociación con múltiples enfermedades autoinmunitarias; sin embargo, la afección hipotálamo-hipofisaria en la enfermedad de Behçet es excepcional. Revisamos los casos publicados y los posibles mecanismos patogénicos de esta asociación hasta ahora no descrita.
We report a case of a patient with longstanding Behçet disease, with neurological symptoms predominantly, who became hospitalized for adrenal insufficiency, caused by isolated deficiency of corticotropin (DAACTH). DAACTH is a typical characteristic of hypophysitis, reported in association with many autoimmune diseases. Nevertheless, hypothalamic-pituitary injury in Behçet disease is exceptional. We review the literature and possible mechanisms of this association until now not reported.
La enfermedad de Behget (EB) es un proceso inflamatorio crónico sistémico de tipo vasculítico y probable origen autoinmunitario, con baja prevalencia en la población general. Las manifestaciones neurológicas ocurren en un 10-20% de los casos; se han descrito varios mecanismos patogénicos que incluyen infiltración focal parenquimatosa, trombosis venosas y, más raramente, meningitis aséptica y vasculitis arterial1,2.
La asociación con endocrinopatías y otras enfermedades autoinmunitarias es infrecuente y generalmente casual; no forma parte de síndromes poliendocrinos3-5. La afección hipotálamo-hipofisaria es excepcional.
Comentamos a continuación el caso de un paciente con enfermedad de Behget y déficit aislado de corticotropina (ACTH), una asociación hasta ahora no descrita.
CASO CLÍNICOVarón de 68 años diagnosticado de enfermedad de Behget en 1978 (a los 38 años), con historia de úlceras orales, genitales y predominio de manifestaciones neurológicas y oculares en forma de ictus isquémicos de repetición y neuritis óptica recurrente. Fue tratado con corticoides a dosis altas, y desarrolló diabetes esteroidea. En 1999 ingresó por pancreatitis litiásica y un cuadro clínico de hipotensión mantenida e hipoglucemias, con alteraciones hidroelectrolíticas (hiponatremia e hiperpotasemia) indicativas de hipoadrenalismo, por lo que fue remitido a nuestro servicio donde se objetivó insuficiencia suprarrenal secundaria a déficit aislado de ACTH (tabla 1), con resonancia magnética (RM) hipofisaria normal. Se instauró inicialmente tratamiento con hidrocortisona intravenosa a dosis de 300mg diarios con resolución del hipoadrenalismo agudo y, luego, tratamiento sustitutivo con hidrocortisona oral, a dosis de 30mg diarios (20mg al desayuno y 10mg a las 20.00), con buena respuesta clínica; 2 años después el paciente abandonó el seguimiento en consultas externas.
Estudio hormonal
| Parámetro (intervalo de referencia) | 1999 | 2000 | 2001 | Enero 2008 | Mayo 2008 |
| Cortisol basal (μg/100ml) | < 0,5 | < 1 | 2,45 | < 1 | < 1 |
| ACTH basal (4-52pg/ml) | 24,1 | 55,9 | 55,05 | 16,34 | 20,24 |
| GH (0-5ng/ml) | 0,42 | 0,06 | 0,72 | 0,32 | |
| IGF-I (80,2-161,4ng/ml) | 88,64 | 135,8 | 32,5 | 189 | |
| IGFBP-3 (3,41-5,46mg/l) | 3,9 | 3,69 | 1,5 | 4,56 | |
| T4L (0,7-2ng/100ml) | 1,66 | 1,5 | 1,56 | ||
| TSH basal (0-3-4,5 μU/ml) | 2,21 | 2,02 | 1,49 | 1,83 | 1,06 |
| FSH basal (1,6-17,5mU/ml) | 4,96 | 8,21 | 3,25 | 4,18 | 9,93 |
| LH basal (1,5-11mU/ml) | 4,7 | 4,29 | 3,37 | 4,73 | 2,88 |
| Prolactina (3–14,7ng/ml) | 27,5 | 11,6 | 11,6 | 8,5 | 8,82 |
| Testosterona basal (3-10ng/ml) | 7,38 | 7,59 | 4,49 | 5,03 | 3,4 |
| Sodio (135–145mEq/l) | 133 | 139 | 134 | 143 | |
| Potasio (3,5-5,1mEq/l) | 4,8 | 3,50 | 5,6 | 4,03 |
ACTH: corticotropina; FSH: folitropina; GH: somatotropina; IGF-1: factor insulinoide similar a la insulina-1; IGFBP-3: proteína de transporte de IGF-3; LH: lutropina; T4L: tiroxina no unida a proteínas; TSH: tirotropina.
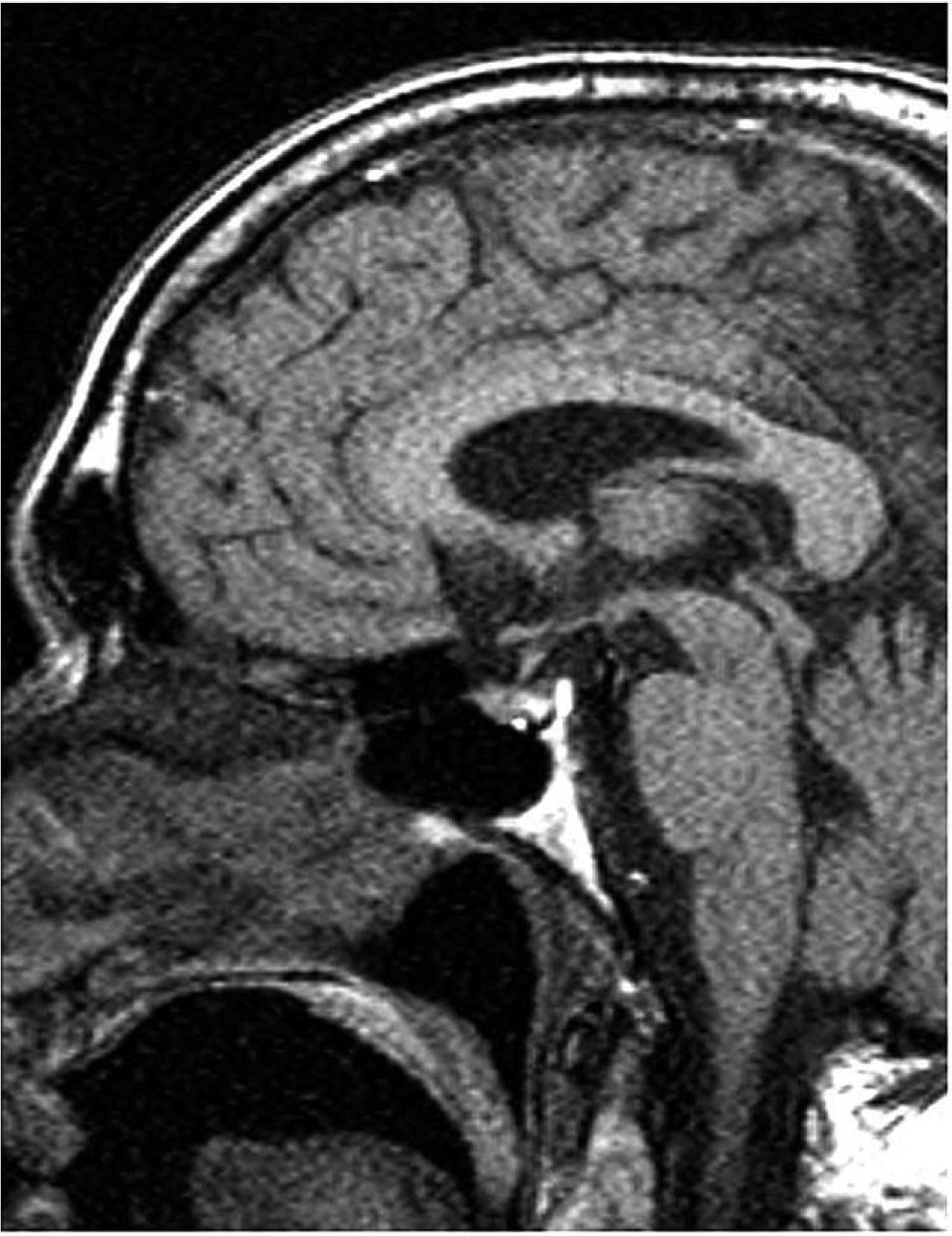
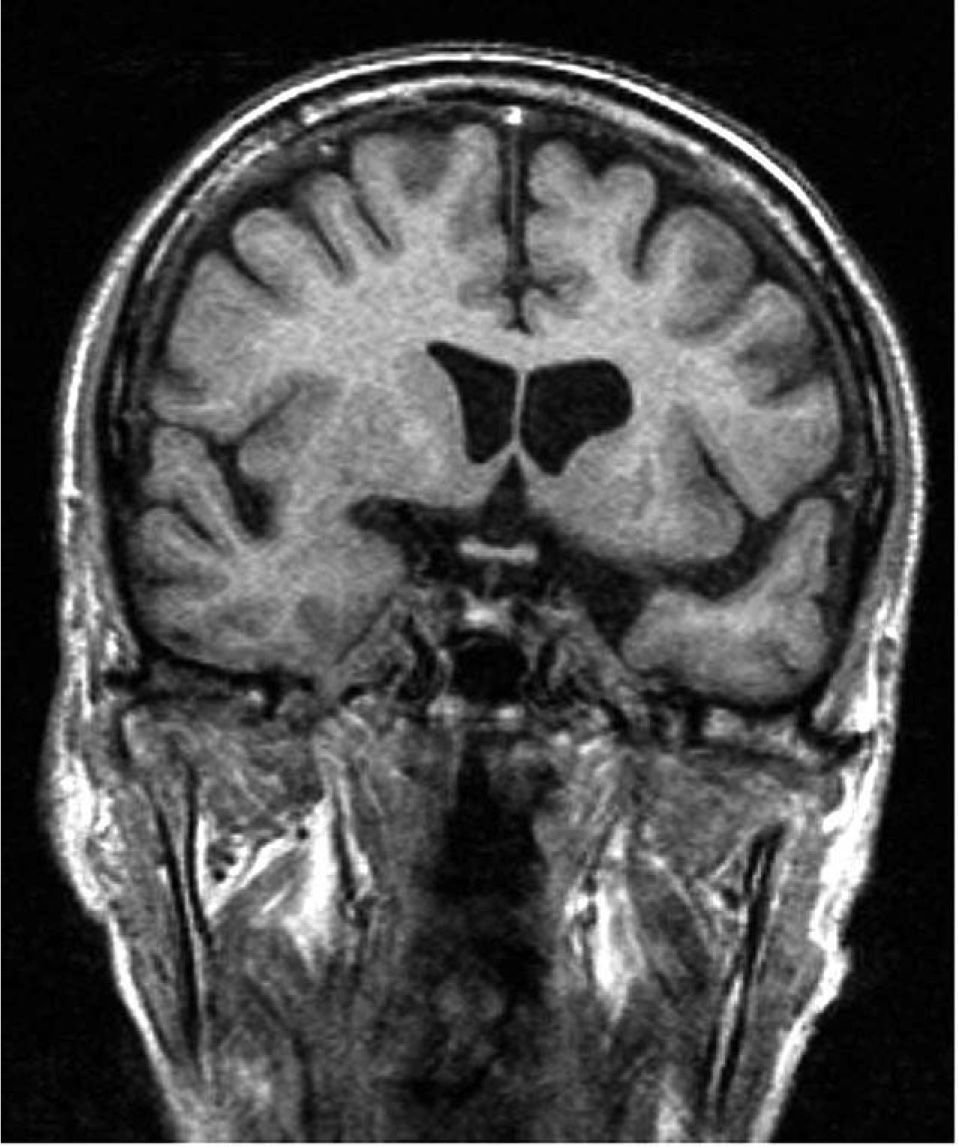
En enero de 2008 fue remitido nuevamente a nuestro servicio por presentar cuadro constitucional y concentraciones séricas de cortisol basal indetectables, estudio solicitado por su médico de cabecera a raíz del antecedente de insuficiencia suprarrenal descrito. El paciente había abandonado el tratamiento sustitutivo esteroideo 5 años antes por temor a las hiperglucemias y, paulatinamente, dejó de precisar antidiabéticos. Su familia relataba deterioro físico y psíquico progresivo muy marcado en los últimos 3 meses, con astenia, anorexia y pérdida de 3kg de peso, sensación perenne de frío, somnolencia y bradipsiquia. No presentaba poliuria ni polidipsia. Días antes del ingreso había sufrido un episodio de desorientación y alteración en la marcha y el habla. En la exploración física destacaba deterioro psicomotor ligero (Minimental test de 18/30), disminución de agudeza visual bilateral y lenguaje disártrico. En la bioquímica realizada en urgencias se obtuvieron los siguientes valores: glucemia de 73mg/dl, sodio de 134mEq/l y potasio 5,60mEq/l, aunque el suero estaba ligeramente hemolizado. El paciente fue tratado con hidrocortisona intravenosa a dosis de 300mg diarios. Ya en planta, en la analítica sistemática no se detectaron alteraciones electrolíticas, con natremia de 140mEq/l, potasemia de 3,65mEq/l y glucemia basal de 93mg/dl. No se evidenciaron alteraciones en la osmolaridad plasmática ni urinaria que indicaran diabetes insípida. El estudio hormonal confirmó el hipoadrenalismo secundario, así como valores bajos de somatotropina (GH) y factor insulinoide similar a la insulina (IGF)-I (tabla 1). Se realizó RM cerebral, sin evidenciarse proceso patológico estructural hipotálamo-hipofisario (figs. 1 y 2). El estudio de autoinmunidad (anticuerpos antitiroglobulina y tiroperoxidasa, anti-GAD, anti-IA2 y anticélulas parietales) fue negativo.


Los potenciales evocados visuales fueron compatibles con neuritis/neuropatía óptica bilateral. Se reinició tratamiento esteroideo con gran mejoría clínica, tanto física como cognitiva, normalización de GH e IGF-I y ligera descompensación glucémica (glucemia basal de 140mg/dl) que se controló con insulinoterapia subcutánea en pauta estándar.
DISCUSIÓNEl déficit aislado de ACTH es una entidad infrecuente, caracterizada por hipocortisolismo secundario, ausencia de defectos estructurales hipofisarios (excepto los característicos de hipofisitis) y secreción normal del resto de las hormonas hipofisarias6, 7. Hay que tener en cuenta que el cortisol está implicado en la regulación de múltiples ejes, por lo que un déficit mantenido puede acompañarse de disminución reversible de GH, hiperprolactinemia y ligero aumento de tirotropina (TSH), que se resuelven con tratamiento esteroideo sustitutivo6,8,9, como ocurrió con nuestro paciente.
Clínicamente puede pasar inadvertido mucho tiempo hasta que un evento estresante desencadene una crisis suprarrenal con síntomas inespecíficos, como astenia, anorexia, adelgazamiento, hipotensión y ortostatismo, asociados a otros hallazgos característicos del hipoadrenalismo, como tendencia a la hipoglucemia, hiponatremia con potasio normal o ligeramente elevado, linfocitosis con eosinofilia y anemia.
La causa más frecuente de déficit aislado de ACTH (DAACTH) es el tratamiento crónico con glucocorticoides. En la infancia suele deberse a alteraciones genéticas. En el adulto, descartando una causa iatrogénica, se relaciona con traumatismos, síndrome de Sheehan, secuela tras radiación y tras cirugía selar; es más frecuente el déficit combinado de varias hormonas hipofisarias6. El DAACTH se describe clásicamente en relación con hipofisitis. Las hipofisitis se deben a un proceso inflamatorio heterogéneo. Se dividen en primarias (linfocítica, granulomatosa, xantomatosa) y secundarias, que son causadas por lesiones de vecindad como tumores o formando parte de enfermedades sistémicas (autoinmunitarias e infecciosas principalmente)10-15. La hipofisitis linfocitaria suele verse en mujeres en el período posparto y se asocia con otras enfermedades autoinmunitarias en un 20-50% de los casos; la más frecuente es la tiroiditis de Hashimoto16-20. La adenohipofisitis se manifiesta por síntomas compresivos o clínica de hipofunción hormonal que, además, es desproporcionada con los hallazgos radiológicos19. Característicamente, el déficit de ACTH es el más frecuente y a veces el único, suele seguirse de disminución de TSH y evoluciona en pocos meses a hipopituitarismo17,19,20.
Se cree que una destrucción autoinmunitaria de las células corticotropas podría ser la causa del déficit de ACTH en algunos pacientes21. Es conocida su asociación con múltiples enfermedades autoinmunitarias (organoespecíficas y sistémicas)6,10-15 y se han detectado anticuerpos antihipofisarios en casi el 70% de los casos comprobados por biopsia19,22.
Las imágenes típicas de la adenohipofisitis son aumento homogéneo de tamaño hipofisario, a veces con extensión supraselar en forma de lengua hacia el hipotálamo. Un dato menos frecuente, pero más específico, es el realce en forma de pico triangular en el diafragma selar16-20. Se ha visto que en estudios dinámicos de RM hay retraso en la captación del contraste en el lóbulo anterior, lo que orienta a vasculopatía hipofisaria17,19,23, y que analíticamente se asocia a déficit hormonales progresivos (GH y TSH), generalmente coexistiendo diabetes insípida central24. Asimismo, pueden presentar engrosamiento del tallo o silla turca vacía19,23,25. Otras veces los estudios de imagen no muestran alteraciones, como en el caso de nuestro paciente.
La afección hipotálamo-hipofisaria asociada a la enfermedad de Behget es excepcional. Tras una revisión de la literatura, hemos encontrado únicamente 6 casos de diabetes insípida central en pacientes con esta entidad26-31. Se han propuesto varios posibles mecanismos patogénicos, como infiltración linfocítica infundibular con hipofisitis secundaria, infiltración meníngea y afección vascular hipofisaria. La presencia de hipopituitarismo es todavía más infrecuente, con dos publicaciones hasta la fecha: un paciente con hipotiroidismo terciario y masa hipotalámica32 y otro con déficit hormonales múltiples en relación con atrofia hipofisaria y vasculitis con oclusión arterial31. No hemos hallado más casos en la búsqueda bibliográfica realizada.
En nuestro paciente, consideramos que el déficit de ACTH estaría relacionado con la afección sistémica por enfermedad de Behget y, por lo tanto, con un origen autoinmunitario. Proponemos como mecanismo patogénico más probable una hipofisitis linfocitaria, aunque con presentación atípica, ya que es infrecuente que no esté afectado el eje tirotropo, así como la lenta evolución que tuvo desde el inicio con hipoadrenalismo. Esto podría explicarse, en parte, por los esteroides administrados, que constituyen el tratamiento de elección en esta enfermedad, junto con otros inmunosupresores7,17-20. De hecho, se han comunicado casos de recuperación tras tratamiento corticoideo33.
