


La enfermedad de Kennedy o atrofia muscular espino-bulbar es un trastorno neurodegenerativo raro de herencia recesiva ligada al cromosoma X que afecta a varones en la edad adulta. Está causado por la expansión repetida de la secuencia citosina-adenosina-guanina en el exón 1 del gen del receptor androgénico localizado en el cromosoma Xq11-12, y se caracteriza por la degeneración progresiva de las neuronas motoras espinales. Desde el punto de vista endocrinológico es común encontrar en estos pacientes datos de hipogonadismo englobados en el síndrome de resistencia androgénica, particularmente la forma parcial.
Se describen 4 casos con presentación clínica neurológica típica de la enfermedad, con debilidad muscular generalizada lentamente progresiva con atrofia y afectación de musculatura bulbar; entre las manifestaciones endocrinológicas observadas la ginecomastia fue la más frecuente. El estudio molecular mostró una expansión anormal del triplete citosina-adenosina-guanina en el gen del receptor androgénico en todos los casos.
Kennedy's disease, also known as bulbospinal muscular atrophy, is a rare, X-linked recessive neurodegenerative disorder affecting adult males. It is caused by expansion of an unstable cytosine-adenine-guanine tandem-repeat in exon 1 of the androgen-receptor gene on chromosome Xq11-12, and is characterized by spinal motor neuron progressive degeneration. Endocrinologically, these patients often have the features of hypogonadism associated to the androgen insensitivity syndrome, particularly its partial forms.
We report 4 cases with the typical neurological presentation, consisting of slowly progressing generalized muscle weakness with atrophy and bulbar muscle involvement; these patients also had several endocrine manifestations; the most common non-neurological manifestation was gynecomastia. In all cases reported, molecular analysis showed an abnormal cytosine-adenine-guanine triplet repeat expansion in the androgen receptor gene.
La enfermedad de Kennedy (EK) o atrofia muscular bulbo espinal es un trastorno neurodegenerativo progresivo de la neurona motora de inicio en la edad adulta1 descrito por primera vez en 1968 y cuya causa genética fue establecida en 19912. La prevalencia descrita de la enfermedad es de 3,3/100.000 habitantes en varones caucásicos europeos3,4 aunque también se considera que existe una gran proporción de casos infradiagnosticados5.
La EK tiene carácter recesivo ligado al cromosoma X, siendo los varones los afectados clínicamente6. Tiene su origen en una mutación dinámica en el gen del receptor de andrógenos (RA) localizado en Xq11-q12, que consiste en una expansión anormal del triplete citosina-adenina-guanina (CAG) en el exón 1 del gen2,3,6–8.
Clínicamente, los pacientes presentan debilidad progresiva en los músculos de las extremidades y a nivel de musculatura bulbar, atrofia, fasciculaciones, temblor o calambres. Las manifestaciones endocrinológicas consisten en hipogonadismo como consecuencia del síndrome de resistencia androgénica3,9–11.
El diagnóstico de la enfermedad se establece mediante la demostración de la presencia de más de 40 tripletes CAG en el gen RA2,3,6.
Se presentan 4 casos de EK con estudio genético confirmado.
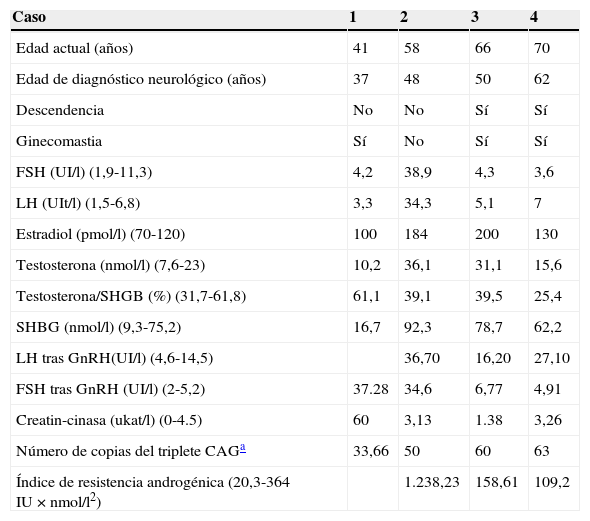
Pacientes y métodosSe describen individualmente los datos clínicos, analíticos y genéticos de 4 pacientes, que se resumen en la tabla 1.
Características clínicas, hormonales y estudio genético de los 4 pacientes
| Caso | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Edad actual (años) | 41 | 58 | 66 | 70 |
| Edad de diagnóstico neurológico (años) | 37 | 48 | 50 | 62 |
| Descendencia | No | No | Sí | Sí |
| Ginecomastia | Sí | No | Sí | Sí |
| FSH (UI/l) (1,9-11,3) | 4,2 | 38,9 | 4,3 | 3,6 |
| LH (UIt/l) (1,5-6,8) | 3,3 | 34,3 | 5,1 | 7 |
| Estradiol (pmol/l) (70-120) | 100 | 184 | 200 | 130 |
| Testosterona (nmol/l) (7,6-23) | 10,2 | 36,1 | 31,1 | 15,6 |
| Testosterona/SHGB (%) (31,7-61,8) | 61,1 | 39,1 | 39,5 | 25,4 |
| SHBG (nmol/l) (9,3-75,2) | 16,7 | 92,3 | 78,7 | 62,2 |
| LH tras GnRH(UI/l) (4,6-14,5) | 36,70 | 16,20 | 27,10 | |
| FSH tras GnRH (UI/l) (2-5,2) | 37.28 | 34,6 | 6,77 | 4,91 |
| Creatin-cinasa (ukat/l) (0-4.5) | 60 | 3,13 | 1.38 | 3,26 |
| Número de copias del triplete CAGa | 33,66 | 50 | 60 | 63 |
| Índice de resistencia androgénica (20,3-364 IU × nmol/l2) | 1.238,23 | 158,61 | 109,2 |
Entre paréntesis se describen los valores dentro del rango de normalidad.
SHBG: globulina fijadora de hormonas sexuales (sex hormone binding globulin); triplete CAG: citosina-adenina-guanina.
Para determinar la FSH, LH y SHBG se utilizó enzimoinmunoanálisis quimioluminiscente no competitivo en fase sólida, con temperatura de incubación de 37°C, tiempo de incubación de 30min, y calibración no lineal con 2 calibradores (analizador Immulite 2000 Siemens). La quimioluminiscencia medida es directamente proporcional a la cantidad de hormona presente en la muestra. Los valores de referencia para FSH y LH son 1,9-11,3 UI/l y 1,5-6,8 UI/l respectivamente; los valores normales para SHBG son 9,3-75,2 nmol/l.
El estradiol y la testosterona total se midieron con la técnica de enzimoinmunoanálisis quimioluminiscente competitivo en fase sólida, con temperatura de incubación 37°C; tiempo de incubación 60min, y calibración no lineal con 2 calibradores. La quimioluminiscencia medida es inversamente proporcional a la cantidad de hormona presente en la muestra. Los valores de referencia para la testosterona son 7,6-23 nmol/l, y para el estradiol, 70-120pmol/l. La normalidad del cociente testosterona/SHGB es 31,7-61,8%.
El producto del valor absoluto de testosterona sérica por el de LH es conocido como índice de resistencia androgénica (androgen sensitivity index [AIS]).
La creatina-cinasa se analizó mediante espectrometría de absorción molecular a 340 y 546nm en el analizador Cobas c701 (Roche Diagnostics). Los valores de referencia son 0-4,5 μKat/L.
El estudio incluyó una prueba dinámica de estimulación de FSH y LH tras administración de 84,6mmol (100mg) de gonadorrelina intravenosa (GnRH). Existen escasos trabajos de referencia que describan valores de normalidad de gonadotrofinas en sujetos con función gonadal normal. Un estudio clásico12 refiere que el incremento de LH tras 100 μg de GnRH es de 2 a 10 veces por encima del normal, mientras que el de FSH es menos evidente. Otro estudio13 describe para varones de entre 18 y 65 años con función gonadal normal valores de normalidad basales de LH de 0,4-6,9 UI/l y de FSH de 0,4-5,8 UI/l mientras que a los 60 min tras estímulo con 100 μg de GnRH son de 4,6-14,5 UI/l y 2-5,2 UI/l, respectivamente.
En todos los casos descritos con sospecha de EK se realizó análisis molecular de ADN mediante la técnica de reacción en cadena de polimerasa (PCR) de la región polimórfica del RA. Se determina el número de tripletes CAG presentes en la zona polimórfica del exón 1 del gen del RA, situado en el cromosoma X. Para ello se realiza una extracción de ADN a partir de sangre total EDTA con columnas Qiagen Mini Spin Blood. Posteriormente, a partir del ADN genómico total se realiza una amplificación por PCR de la región polimórfica del gen RA. La amplificación se realiza con Taq polimerasa (Ecotaq) según el programa 95¿-5′+35 X (95¿-45″+68¿-40″+72¿-1′ 10″)+(72¿-10′), en un termociclador Applied Biosystems GeneAmp 2700.
Esta PCR es analizada mediante electroforesis en un gel de agarosa al 2%. El número de tripletes presente en la muestra se determina de acuerdo con el tamaño, siendo los valores inferiores a 35 tripletes (hasta 300 pb) considerados normales y los superiores a 36, patológicos.
Caso 1Paciente varón de 41 años, sin antecedentes patológicos (AP) de interés salvo queratoplastia. Diagnosticado a los 37 años por clínica neurológica consistente en debilidad proximal en extremidades inferiores (EEII); la exploración objetivó paresia proximal junto con arreflexia, atrofia lingual y debilidad facial progresiva. En el momento actual mantiene autonomía con libre funcionalidad manipulativa y capacidad de marcha libre.
Presentaba ginecomastia y disminución del vello corporal desde la pubertad. No refería cambios en la voz ni a nivel de musculatura, pero refería disminución de la libido y potencia sexual de 3 años de evolución. No había logrado tener descendencia aunque nunca se había practicado estudio de fertilidad.
En el estudio genético presentó 60 repeticiones del triplete CAG, lo que corresponde a un rango considerado patológico (de 36 a 88 repeticiones de CAG).
En el estudio de familiares de primer grado se diagnosticó a la madre como portadora de la mutación, mientras que el abuelo materno había sufrido un cuadro neurológico similar pero no había sido estudiado.
Exploración física: peso (P) 84kg; talla (T) 1,69m; indice de masa corporal (IMC) 29,4kg/m2; tensión arterial 114/62mmHg. Sin bocio. No presentaba proporciones eunucoides, existía disminución de vello corporal y ginecomastia bilateral de predominio izquierdo. Volumen testicular: teste derecho (TD) 12 cc y teste izquierdo (TI) 10 cc.
Caso 2Paciente varón de 58 años, con AP de hipertensión arterial (HTA) en tratamiento farmacológico, dislipidemia leve sin tratamiento y litiasis renal. Diagnosticado a los 48 años a raíz de clínica neurológica en forma de calambres, astenia, fatigabilidad y debilidad generalizada.
Había presentado desarrollo puberal normal sin aparición de ginecomastia. Refería alopecia androgénica sin cambios en la frecuencia del afeitado ni disminución de vello corporal. Explicaba disminución progresiva de la potencia de la voz y del volumen de los testículos en los últimos años, así como disminución de la libido en los últimos 15 años. Describía impotencia sexual desde el inicio de la clínica neurológica.
En el estudio genético presentó 50 repeticiones del triplete CAG.
No se había practicado estudio genético en miembros familiares de primer grado y no había tenido hijos a pesar de no utilizar medidas anticonceptivas con pareja estable en 35 años. Nunca se había realizado estudio de fertilidad.
Exploración física: P 76kg;T 1,75m; IMC 26,3kg/m2; TA 150/95mmHg. Sin bocio. No ginecomastia. Vello facial y corporal de distribución masculina. Tamaño testicular: TD/TI 20/12 cc.
Caso 3Paciente varón de 66 años, con AP de HTA en tratamiento farmacológico. Inicio de clínica neurológica a los 52 años con debilidad y fatigabilidad de EEII con fasciculaciones y frecuentes caídas. Dichos síntomas habían progresado hasta dificultar la deambulación, por lo que el paciente actualmente precisaba uso de silla de ruedas.
Presentaba ginecomastia bilateral desde la infancia sin cambio tras inicio de los síntomas neurológicos. Asimismo, había sufrido retraso en la aparición de caracteres sexuales secundarios; consultó en la adolescencia sin alcanzar un diagnóstico preciso y completó tardíamente el desarrollo puberal. De forma paralela al inicio de síntomas neurológicos refería pérdida en la potencia de la voz y disfunción eréctil, sin disminución de la libido.
En el estudio genético presentó 60 repeticiones del triplete CAG.
No se había practicado estudio genético en padres ni hermanos, tenía 2 hijas que fueron estudiadas y eran portadoras de la mutación; una de ellas tenía 2 hijos varones también estudiados en los que se había descartado la alteración genética.
Exploración física: P 81kg;T 1,75m; IMC 26,4kg/m2; TA 140/50mmHg. Sin bocio. Presentaba ginecomastia leve con predominio de mama izquierda. Distribución masculina de vello facial y corporal. Tamaño testicular: TD/TI 20/15-20 cc.
Caso 4Paciente varón de 70 años, con AP de HTA y dislipidemia en tratamiento farmacológico, y síndrome de apnea obstructiva del sueño en tratamiento con presión positiva continua en las vías respiratorias. Había sido diagnosticado a los 62 años por clínica neurológica en forma de debilidad distal en extremidades superiores y parestesias. La exploración mostró arreflexia, atrofia de musculatura intrínseca de manos y apalestesia distal en EEII. Presentaba desde el diagnóstico cambios en la potencia de la voz y ligera disfagia.
Describía desarrollo puberal normal sin ginecomastia ni otros datos de hipogonadismo.
A los 64 años había sido diagnosticado de neoplasia de próstata que fue tratada con radioterapia, desde entonces el paciente explicaba disminución de libido no presente antes del tratamiento específico.
En el estudio genético presentó 63 repeticiones del triplete CAG.
No se había practicado estudio genético en familiares de primer grado. Tenía 5 hijos (3 varones y 2 mujeres) y una nieta, en todos ellos se había descartado la mutación.
Exploración física: P 73,6kg;T 1,64m; IMC 27,4kg/m2; TA 120/60. Sin bocio. Mínima ginecomastia derecha. Distribución masculina de vello facial y corporal. Exploración testicular: TD/TI 20/20 cc. Hernia inguinal izquierda y cicatriz inguinal por hernia inguinal derecha intervenida.
DiscusiónLa EK es un trastorno neurodegenerativo asociado a expansión anormal del triplete CAG en el gen que codifica para el RA5,14.
Se trata de una enfermedad de presentación tardía; la edad promedio de inicio de los síntomas en nuestra serie es 49,3 años (37-62 años), similar a lo descrito en la literatura con rangos entre la segunda y sexta décadas de la vida3,4,7,9,15,16. Se describen casos de inicio en la etapa adolescente que cursan inicialmente con signos de hipogonadismo como ginecomastia, microorquidia, oligospermia o azoospermia9.
La afectación neurológica comprende los característicos signos de afectación de la neurona motora inferior, tales como debilidad muscular proximal o distal, atrofia muscular, fasciculaciones a nivel facial, perioral y de extremidades, disartria o disfagia17. Los síntomas iniciales incluyen temblor, calambres y debilidad muscular17. No están descritos signos de enfermedad de la neurona motora superior como hiperreflexia o espasticidad3.
El espectro clínico neurológico es muy amplio. Los datos fenotípicos pueden variar desde una alteración analítica aislada, como puede ser la elevación de la concentración sérica de creatin-cinasas (CK) secundaria a la atrofia muscular, hasta las formas más graves con afectación de la musculatura bulbar, que pueden precisar ventilación artificial y colocación de sondas de gastrostomía para alimentación3,6.
En nuestra serie el síntoma inicial principal que motivó la consulta fue la debilidad muscular y fatigabilidad de EEII, con seria dificultad para la marcha en uno de los casos. Los síntomas neurológicos predominantes fueron, junto a esta debilidad de EEII, los calambres, la atrofia lingual y el compromiso bulbar variable, reconocidos como hallazgos característicos de la enfermedad3. Uno de los casos presentó hipoestesia distal dolorosa paralela a los síntomas de debilidad muscular, descrito en menos de la mitad de los casos en otros estudios1,3,18.
Los hallazgos endocrinológicos de nuestros pacientes se caracterizaron por la presencia de ginecomastia, disminución de vello corporal, reducción del tamaño testicular, disminución de la libido e impotencia sexual3,9,19,20.
Dos de los casos presentaban ginecomastia bilateral desde la infancia y la pubertad lo que se aproxima a la frecuencia del 70% descrita por Dejager et al.9; se considera que se trata de la manifestación endocrinológica más frecuente3,20. Algunos pacientes pueden desarrollar estos síntomas endocrinológicos y, en particular, la ginecomastia, antes incluso de la clínica neurológica9.
Dos de los casos presentaron disminución de libido y disfunción eréctil con una evolución paralela a los síntomas neurológicos, mientras que en los otros 2 casos la clínica neurológica tuvo una aparición posterior a las alteraciones endocrinológicas. En 2 casos se objetivó un tamaño testicular reducido sin encontrar signos de ambigüedad sexual como hipospadias o micropene.
Los datos del estudio hormonal de nuestra serie de pacientes se resumen en la tabla 1. El primer paciente presenta un estudio hormonal dentro de la normalidad. En 2 de los casos encontramos incrementada la concentración de testosterona total sérica (T), así como la SHGB, con una fracción libre sérica de testosterona disminuida9. En la literatura está descrito que el nivel sérico de T puede aparecer normal o elevado9,16,21–23, con una concentración alta incluso a edades avanzadas16; puede existir una correlación positiva entre la T y la SHGB, que puede estar inapropiadamente aumentada3,9,20. En cuanto al nivel sérico de estrógenos (E2) aparece aumentado en los 3 últimos casos. Este incremento puede deberse a la aromatización de testosterona a estrógenos2,9,22.
Las gonadotrofinas se hallan aumentadas en el segundo caso, mientras que la LH está ligeramente elevada en el cuarto caso. Tras practicar la prueba de estimulación con GnRH en los 3 últimos pacientes se comprobó una hiperrespuesta de la LH; por otro lado, el segundo y tercer pacientes presentaban también hiperrespuesta de la FSH, que en cambio no se observaba en el último caso. Los valores de LH referidos en la literatura pueden ser elevados o no suprimidos; la respuesta de LH al estímulo con GnRH puede ser exagerada a pesar del nivel sérico normal de testosterona, reflejo de la resistencia parcial androgénica9.
La elevada concentración sérica de LH es el resultado de la resistencia de hipotálamo e hipófisis al mecanismo de feed-back negativo por los esteroides sexuales, debido a la resistencia androgénica. Consecuentemente, la LH estimula una mayor producción de testosterona y estradiol por las células de Leydig11,24. Los niveles séricos de FSH en 3 de nuestros 4 pacientes son normales, coincidiendo con lo publicado22,23.
El AIS, resultado del producto de la multiplicación de LH por testosterona, se ha planteado como un buen indicador de resistencia androgénica, que refleja la alteración del feed-back negativo sobre el eje hipotálamo-pituitario-testicular. Se considera que puede ser un marcador útil para identificar a pacientes de riesgo para formas leves de resistencia androgénica causadas por mutaciones en RA. Al calcular este índice en nuestra serie se obtiene una media de 384,93 IU × nmol/l2. En un estudio previo con 22 pacientes afectos por EK confirmada, la media de AIS fue de 166 IU × nmol/l2 (rango 20,3-364 IU × nmol/l2)9, superior a la obtenida en un grupo control de hombres sanos fértiles (media 36 IU × nmol/l2, rango 3,5-88 IU × nmol/l2) o de otros grupos control extraídos de estudios más antiguos (54 IU × nmol/l2, rango 6,7-138,7 IU × nmol/l2)23.
Dejager et al.9 encuentran una correlación significativa entre el tamaño de la expansión del triplete CAG, AIS y la SHGB9; no hemos observado dicha correlación en nuestra serie.
En pacientes clínicamente afectos también se han descrito niveles de CK elevados. En nuestra serie el primer caso presentaba niveles de CK 8 veces superiores a la normalidad19.
El cuadro endocrinológico de la EK se incluye en el síndrome de resistencia parcial androgénica (PAIS acrónimo del inglés partial androgen insensitivity syndrome), con pérdida parcial de caracteres sexuales secundarios como ginecomastia, disminución de vello facial y frecuencia de afeitado, atrofia testicular, disminución de la libido, disfunción eréctil y oligospermia/azoospermia que causa alteraciones de la fertilidad11,25.
El RA es un factor de transcripción hormonal nuclear responsable de la acción androgénica en tejidos diana como vesículas seminales o próstata, entre otros, con un papel central en la virilización del feto masculino y la espermatogénesis. También se expresa en neuronas motoras de nervios espinales y craneales5. Se localiza en el cromosoma Xq11-12 y está formado por 8 exones3,16,26. El primer exón, que codifica la región N-terminal con un importante papel en la transactivación, engloba la secuencia de nucleótidos CAG que, a su vez, codifica el tramo de poliglutaminas en la correspondiente proteína del RA5,27,28. Las variaciones en esta cadena de poliglutaminas se han asociado con la EK2,8.
Los mecanismos que subyacen a la neurodegeneración en la EK no están totalmente aclarados. Es probable que la expansión de la repetición de triplete en el gen RA dé lugar a proteínas disfuncionales con secuencias de poliglutamina largas, que, cuando se translocan al núcleo, producen inclusiones nucleares de poliglutamina y la desregulación de las transcripciones29. La expresión de RA es particularmente abundante en las neuronas motoras, donde los andrógenos juegan un papel importante en la regeneración axonal30.
Mientras que los individuos normales presentan un rango de 9 a 36 copias de triplete CAG3,20 los hombres afectos y las mujeres portadoras tienen una expansión patológica de dicha región, de 40 a 62 repeticiones3,16,26 que causa una ganancia de función de la proteína y conduce a neurotoxicidad3,17,26. Datos experimentales y clínicos apoyan la hipótesis de la ganancia de funciones neurotóxicas como base molecular de la enfermedad31.
En diferentes estudios se ha descrito una correlación positiva entre el tamaño de la expansión y la gravedad de la enfermedad; también se ha correlacionado una menor edad de aparición de la ginecomastia con un mayor grado de resistencia androgénica9. Varias revisiones coinciden en una correlación negativa entre el tamaño de la expansión y la edad de aparición de la enfermedad3,9,16,32,33. Asimismo, otras revisiones de la literatura establecen una relación directa con la tasa de progresión23 que, sin embargo, no se confirma en otros estudios16. En nuestra serie, el paciente con mayor número de copias no se diagnosticó a una edad más precoz que el resto de los casos, ni presentaba una peor evolución de la enfermedad.
Las mutaciones que afectan al gen RA se pueden clasificar en 2 categorías principales: las mutaciones que originan la expansión del triplete CAG (ganancia de función neurotóxica del RA portador de la región poliglutamina elongada) y que no afectan a la diferenciación sexual pero causan EK, y las mutaciones que llevan a una pérdida de funcionalidad del receptor28. Estas últimas producen trastornos de diferenciación sexual en los individuos con genotipo 46 XY, agrupados en el síndrome de resistencia androgénica (SRA)5. Dentro del SRA existe una subclasificación en 2 fenotipos en función de la gravedad de resistencia androgénica: resistencia androgénica completa (complete androgen insensitivity syndrome o CAIS) y resistencia androgénica parcial (partial androgen insensitivity syndrome o PAIS)11,34,35.
Los individuos con CAIS se caracterizan por tener un fenotipo femenino con genitales externos femeninos, vagina corta y ausencia de estructuras derivadas de los conductos de Wolff como útero y trompas de Falopio; asimismo, presentan correcto desarrollo mamario en la pubertad con ausencia de vello púbico o axilar5,10,34–36.
El PAIS es una forma incompleta de SRA con un amplio espectro de presentación clínica que varía desde individuos con genitales externos de apariencia predominantemente femenina a individuos con genitales ambiguos o con un fenotipo fundamentalmente masculino con micropene, hipospadias perineales o criptorquidia (fenotipo incluido anteriormente como síndrome de Reifenstein)5,10,22,34,35.
En la literatura también se describe una tercera variante, el síndrome de resistencia androgénica leve (mild androgen insensitivity syndrome o MAIS) que engloba varones sanos con síntomas de infravirilización, con varios grados de ginecomastia, escaso vello sexual e impotencia. La espermatogénesis en este grupo puede estar alterada o no; por tanto, dichos individuos podrían tener diferentes grados de fertilidad10,11,34. Para algunos autores la EK podría englobarse dentro de este subtipo10.
Los 4 casos aquí presentados podrían considerarse como de PAIS leves o MAIS; incluso 2 de ellos han tenido descendencia. Sin embargo, una limitación de nuestro estudio es que no se dispone de la confirmación genética de SRA de los pacientes presentados.
El RA se expresa en la mayoría de neoplasias de próstata37; dicha asociación indica que el RA puede ser un predictor genético de susceptibilidad individual frente al cáncer de prostata37. En este tipo de neoplasia dependiente de andrógenos, la progresión, crecimiento celular y la supervivencia dependen de la regulación del RA33.
Estudios clínicos epidemiológicos han establecido que la reducción en la expansión del triplete CAG a menos de 22 copias se correlaciona de forma directa con el diagnóstico del cáncer de próstata a edades más precoces, con un riesgo incrementado y una mayor predisposición para un estadio más avanzado de esta neoplasia33,38,39.
Un paciente afecto de EK en nuestra serie fue diagnosticado posteriormente de neoplasia de próstata y, tras estudio molecular, se objetivó un total de 63 copias para el triple CAG. Este resultado contrasta con lo descrito previamente para la neoplasia de próstata aislada.
El diagnóstico de la EK se establece a través de la historia clínica, examen físico, análisis hormonal, herramientas de diagnóstico neurológico como potenciales evocados, electromiografía, estimulación magnética transcraneal y estudios genéticos6. El diagnóstico de confirmación es la demostración a través del estudio genético de la expansión de la repetición del triplete CAG en el gen RA3. En nuestra serie de casos se confirmó la expansión del triplete CAG con más de 36 copias en todos ellos.
La gran mayoría de pacientes tienen una historia familiar positiva pero aproximadamente un tercio de los diagnosticados desconocen datos de otros probables familiares afectos17. Los análisis genéticos permiten un diagnóstico de precisión con una base individual y consejo genético como parte del tratamiento3,6.
No existe tratamiento establecido de la EK, salvo el manejo sintomático3,5,6. Los pacientes pueden recibir vitamina E, complejo vitamínico B o fisioterapia muscular como parte del tratamiento3,6. La sustitución hormonal con testosterona o análogos podrían incluso deteriorar de forma reversible los síntomas3,5,40. Se han realizado ensayos basados en el déficit androgénico en ratones mediante fármacos como el agonista de GnRH leuprorelina, o el inhibidor de la 5-alfa reductasa dutasteride41; se previene así la translocación nuclear de RA aberrante42,43, lo que protege de la acumulación tóxica de la proteína mutante del RA, y logra suprimir las manifestaciones de deterioro neuromuscular. Sin embargo, ensayos clínicos aleatorizados controlados con placebo no han demostrado efectos significativos sobre determinadas funciones neurológicas como la fuerza muscular o la deglución40,44.
Únicamente algunos pacientes requieren soporte ventilatorio en fases avanzadas de la enfermedad, pero la esperanza de vida en general está ligeramente comprometida3.
ConclusiónEn conclusión, presentamos 4 casos de EK confirmados por estudio molecular, con un cuadro clínico neurológico y endocrinológico acorde con otras series. Debe considerarse el diagnóstico de esta entidad en todo varón adulto con debilidad muscular progresiva y alteraciones endocrinas asociadas, con o sin antecedentes familiares.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
