


La neurofibromatosis tipo 1 (NF-1) o enfermedad de von Recklinhousen es uno de los síndromes clásicamente asociados al feocromocitoma. Es una enfermedad multisistémica que afecta principalmente a la piel y al sistema nervioso, de herencia autosómica dominante y expresividad clínica muy variable incluso en miembros de la misma familia. Hasta el 50% son mutaciones de novo y carecen de antecedentes familiares1.
El gen de la neurofibromatosis se encuentra en el cromosoma 17 y codifica la neurofibromina, proteína implicada en la regulación de p21 ras que actúa como supresor tumoral. Por tanto, su pérdida de función permitiría la proliferación celular descontrolada y la aparición de diversos tumores2.
El diagnóstico de la NF-1 es clínico y se establece cuando están presentes al menos 2 de los siguientes criterios3: 1) 6 o más manchas café con leche, 2) 2 o más neurofibromas cutáneos o un neurofibroma plexiforme, 3) pecosidad axilar, 4) nódulos de Lisch, 5) glioma óptico, 6) displasia ósea y 7) familiar de primer grado con NF-1. El 97% de los pacientes cumplen criterios a los 8 años y el 100% a los 20 años. El diagnóstico genético es posible en más del 95% de los pacientes1 pero es técnicamente complejo y no predice la aparición de complicaciones ya que en general no hay correlación fenotipo-genotipo. Por tanto, no está indicado realizarlo de forma rutinaria4.
Entre las múltiples manifestaciones de la NF-1 se encuentra el feocromocitoma, que aparece en el 1-5% de los pacientes5. El feocromocitoma familiar en el contexto de la neurofibromatosis es raro con pocos casos publicados6. Se presentan 2 casos de feocromocitoma en madre e hija afectas de NF-1 y se revisan los aspectos más relevantes de esta asociación.
Caso 1: mujer sin antecedentes personales ni familiares de interés, salvo madre hipertensa fallecida a los 54 años por ictus. Padre y 5 hermanos sanos. Diagnosticada de NF-1 a los 36 años tras una biopsia de tumor inguinal compatible con neurofibroma. En la exploración física presentaba más de 6 manchas café con leche, pequeñas tumoraciones en dedos de ambas manos y nódulos de Lisch en la evaluación oftalmológica. Tras el diagnóstico sus 4 hijos fueron evaluados cumpliendo todos ellos criterios de NF-1.
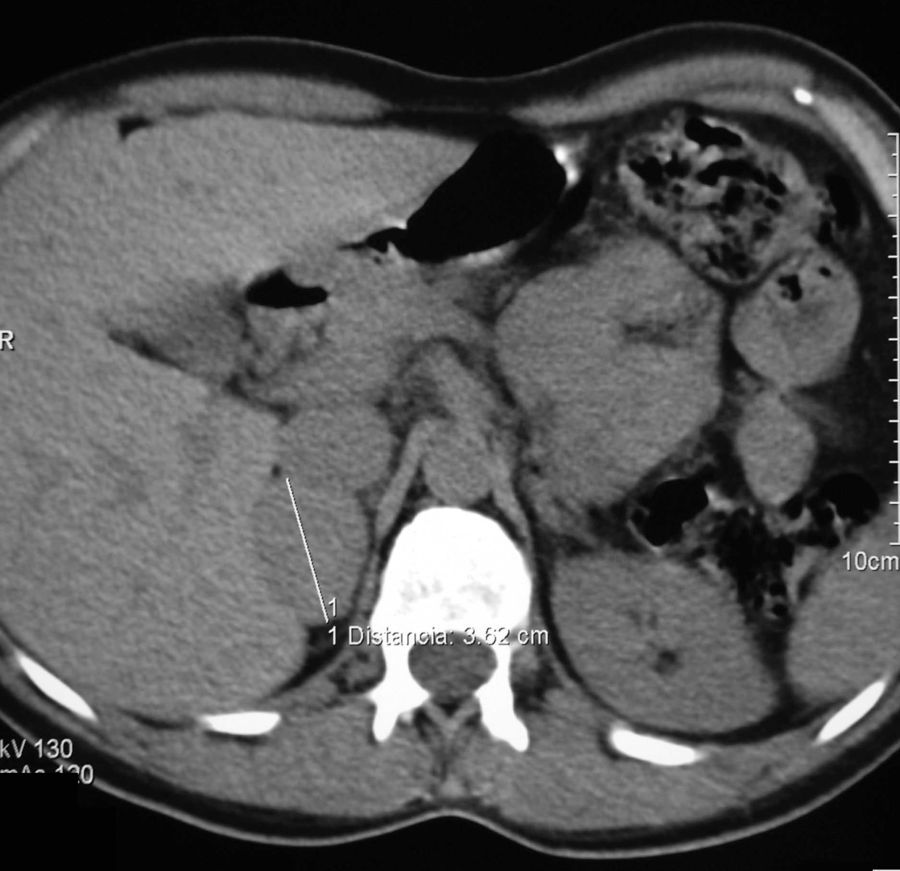
A los 40 años fue estudiada por dispepsia y diarreas, visualizándose en la ecografía abdominal un nódulo adrenal derecho. Refería sudoración e intolerancia al calor en los últimos 2 meses y la TA en consulta fue de 158/92mmHg. Analítica general sin alteraciones. Se solicitaron catecolaminas y metanefrinas urinarias con los siguientes resultados: adrenalina: 117mcg/24h (VN: 0,5-20mcg/24h), noradrenalina: 424mcg/24h (VN: 14-80mcg/24h), metanefrina: 1.386mcg/24h (VN: 86-320mcg/24h), y normetanefrina: 1.720mcg/24h (VN: 129-400mcg/24h). La TC abdominal confirmó un nódulo adrenal derecho de 3,5cm, homogéneo, de 44UH (fig. 1). La gammagrafía con meta-I-bencil-guanidina (MIBG) mostró captación en la suprarrenal derecha. Se intervino por vía laparoscópica tras alfa bloqueo farmacológico con 10mg/d de fenoxibenzamida. La anatomía patológica fue compatible con feocromocitoma encapsulado.
.")
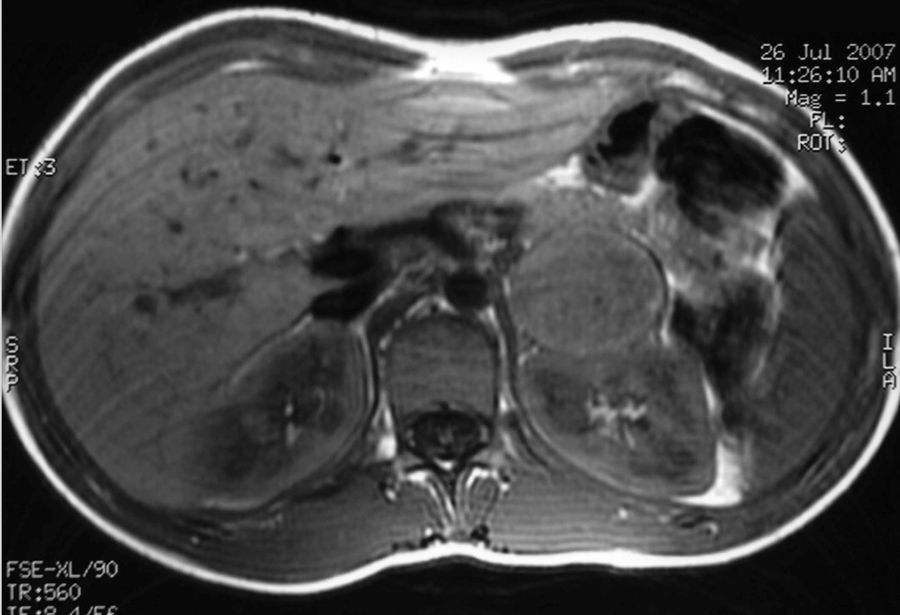
Caso 2: paciente diagnosticada de NF-1 a los 9 años tras el diagnóstico materno (caso 1). A los 15 años consultó por episodios de cefaleas y palpitaciones. En la exploración física destacaban más de 6 manchas café con leche y una TA de 154/83mmHg. Analítica general con glucemia basal de 128mg/dl sin otras alteraciones y TC craneal y evaluación cardiológica normales. Se solicitaron catecolaminas y metanefrinas urinarias con los siguientes resultados: adrenalina: 150mcg/24h (VN: 0,5-20mcg/24h), noradrenalina: 713mcg/24h (VN: 14-80mcg/24h), metanefrina: 1.757mcg/24h (VN: 86-320mcg/24h), normetanefrina: 3.967mcg/24h (VN: 129-400mcg/24h). La RMN describió un nódulo adrenal izquierdo de 4×4×3,8cm hiperintenso en T2, heterogéneo y con zonas necróticas (fig. 2). La MIBG mostró una hiperfijación suprarrenal izquierda. La paciente fue intervenida por vía laparoscópica tras un tratamiento con fenoxibenzamida a dosis de 20mg/12h, confirmándose en la anatomía patológica el diagnóstico de feocromocitoma.
.")
Tras la intervención ambas han permanecido asintomáticas y normotensas, y las catecolaminas y metanefrinas urinarias anuales han sido normales. Se solicitaron catecolaminas y metanefrinas en los otros hijos del caso 1 con resultados normales.
El feocromocitoma aparece en el 1-5% de los pacientes con NF-15. A diferencia del asociado a otros síndromes genéticos, sus características clínicas son similares a las del feocromocitoma esporádico7. La mayoría aparecen en adultos con una edad media de 42 años, aunque el rango es variable (1,5 a 75 años)8. Generalmente, presentan hipertensión arterial (HTA) y/o una clínica compatible, aunque hasta el 22% pueden estar asintomáticos y normotensos8. La localización es mayoritariamente adrenal unifocal. El 10-20% son multifocales7–10 y el 6% extraadrenales5,8. Hasta el 12% son malignos7,8. Nuestros casos fueron adrenales unifocales con una clínica asociada e HTA. En el caso 1 el diagnóstico se realizó tras hallarse incidentalmente una masa adrenal, pero la paciente presentaba una clínica compatible e HTA. El diagnóstico de NF-1 suele establecerse en la infancia1, por lo que es poco probable que los pacientes se presenten como feocromocitomas aparentemente esporádicos. No obstante, existen fenotipos leves que han podido pasar desapercibidos, por lo que deben buscarse signos de NF-1 en feocromocitomas aparentemente esporádicos.
El rango de mutaciones detectadas en pacientes con NF-1 y feocromocitoma es muy amplio y no se conocen mutaciones específicas que favorezcan su aparición o condicionen su presentación o malignidad. Un estudio de Neuman et al. intentó describir el espectro mutacional en 37 pacientes con feocromocitoma y NF-1. Hallaron 36 mutaciones diferentes distribuidas a lo largo de todo el gen sin correlaciones significativas con «hot spots» específicos5.
Las pruebas diagnósticas bioquímicas y de imagen no difieren de las empleadas en el feocromocitoma esporádico. Los feocromocitomas asociados a NF-1 son predominantemente secretores de adrenalina y presentan cifras elevadas de metanefrina y normetanefrina en plasma y orina9.
El tratamiento es quirúrgico, preferiblemente por laparoscopia, tras la preparación con alfa bloqueantes. El seguimiento posterior no difiere del feocromocitoma esporádico. Se recomienda una determinación anual de catecolaminas y metanefrinas que en nuestras pacientes han sido normales.
Dada la baja frecuencia de feocromocitomas en pacientes con NF-1 no está indicado el screening bioquímico rutinario8. Las guías clínicas recomiendan, en niños y adultos con fenotipos leves asintomáticos, al menos una revisión anual con anamnesis de síntomas típicos y una toma de la TA1–3. En pacientes con clínica o HTA deben determinarse catecolaminas y metanefrinas en sangre y/o orina. La HTA es común en pacientes con NF-1 y, aunque las causas más frecuentes son la HTA esencial en adultos y la estenosis de arteria renal en niños, debe descartarse siempre la presencia de un feocromocitoma2. También se recomienda descartarlo en pacientes que se plantean la gestación o vayan a someterse a algún procedimiento médico8.
Sin embargo, el screening rutinario podría aumentar la prevalencia del feocromocitoma hasta el 14,6%, como ha reportado un estudio reciente10, ya que un porcentaje de pacientes se encuentran asintomáticos y normotensos.
No existen recomendaciones específicas para el manejo de los familiares de los pacientes con feocromocitoma asociado a la NF-1. Tras el diagnóstico del caso 2 se determinaron catecolaminas y metanefrinas urinarias en el resto de hermanos afectos con resultados normales. Ogawa et al. también lo consideraron en un caso de feocromocitoma familiar similar al nuestro6. Dado que con la misma mutación la variabilidad clínica intrafamiliar es menor que la interfamiliar, cuando se diagnostica feocromocitoma en 2 miembros de una familia con NF-1 parece prudente realizar screening bioquímico del resto de familiares afectos.
