






La obesidad se asocia con importantes anomalías en la función endocrina. La hiper insulinemia y la resistencia a la insulina son las dos alteraciones mejor conocidas, aunque sus mecanismos y su significado clínico no están claros. El tejido adiposo se considera un órgano endocrino con secreción hormonal; el aumento en la secreción de leptina, una señal de saciedad, por el adipocito es una alteración característica. En la obesidad hay una disminución en la secreción de hormona de crecimiento; esta alteración en la función somatotropa de la obesidad es funcional y se puede revertir en determinadas circunstancias. El mecanismo fisiopatológico responsable de la hiposecreción de GH en la obesidad es probablemente multifactorial. Existen muchos datos que sugieren que un estado crónico de hipersecreción de somatostatina resulta en una inhibición de la liberación de GH; el aumento de los ácidos grasos libres probablemente contribuye a esta alteración, así como un déficit en la secreción de ghrelina. En mujeres, la obesidad abdominal se asocia a hiperandrogenismo y a niveles disminuidos de proteína transportadora de hormonas sexuales. Los hombres obesos tienen niveles de testosterona y concentraciones de gonadotropinas disminuidos, especialmente en los casos de obesidad mórbida. La obesidad se asocia con un aumento en la tasa de producción de cortisol, que se compensa con un aumento del aclaramiento del mismo, lo cual resulta en niveles plasmáticos de cortisol libre que no se modifican con el aumento del peso corporal. Ghrelina es el único factor orexígeno circulante conocido y se ha visto que se encuentra disminuido en humanos obesos. En la obesidad hay también una tendencia a aumentar las concentraciones de TSH y T3 libre.
Obesity is associated to significant disturbances in endocrine function. Hyper insulinemia and insulin resistance are the best known changes in obesity, but their mechanisms and clinical significance are not clearly established. Adipose tissue is considered to be a hormone-secreting endocrine organ; and increased leptin secretion from the adipocyte, a satiety signal, is a well-established endocrine change in obesity. In obesity there is a decreased GH secretion. Impairment of somatotropic function in obesity is functional and may be reversed in certain circumstances. The pathophysiological mechanism responsible for low GH secretion in obesity is probably multifactorial. There are many data suggesting that a chronic state of somatostatin hypersecretion results in inhibition of GH release. Increased FFA levels, as well as a deficient ghrelin secretion, probably contribute to the impaired GH secretion. In women, abdominal obesity is associated to hyperandrogenism and low sex hormone-binding globulin levels. Obese men, particularly those with morbid obesity, have decreased testosterone and gonadotropin levels. Obesity is associated to an increased cortisol production rate, which is compensated for by a higher cortisol clearance, resulting in plasma free cortisol levels that do not change when body weight increases. Ghrelin is the only known circulating orexigenic factor, and has been found to be decreased in obese people. In obesity there is also a trend to increased TSH and free T3 levels.
La obesidad se define como un exceso de grasa corporal. Su manejo clínico es complejo y frecuentemente con pobres resultados1. La obesidad abdominal, u obesidad central, refleja la cantidad de grasa visceral y se relaciona de manera directa con la resistencia a la insulina y los eventos cardiovasculares.
Hay una epidemia de sobrepeso y obesidad en todo el mundo. La prevalencia de exceso de peso está creciendo rápidamente a lo largo de EE.UU. y alrededor del 65% de la población adulta tiene sobrepeso u obesidad2. Si comparamos el periodo 1976-19803 con 1999-20002, la prevalencia de sobrepeso ha aumentado en un 40% (del 46 al 64,5%) y la prevalencia de obesidad se ha incrementado en un 110% (del 14,5 al 30,5%). España no es ajena a esta epidemia y la prevalencia de obesidad y sobrepeso en nuestro medio ha aumentado de forma dramática4, siendo en algunos estudios la prevalencia de exceso de peso (obesidad más sobrepeso) de alrededor del 65% y de obesidad del 25%5.
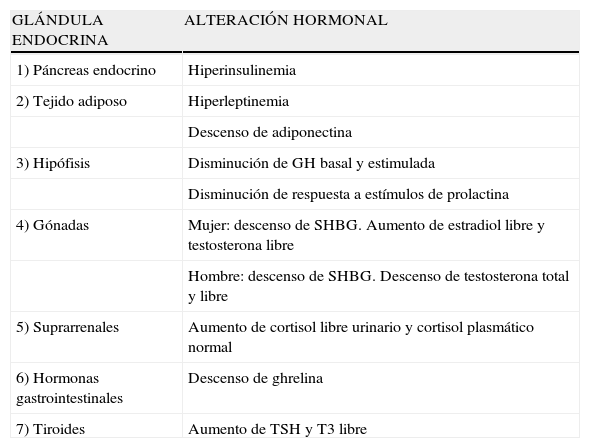
La obesidad es una enfermedad crónica al igual que la hipertensión o la diabetes. La etiología de la obesidad es un desequilibrio entre la energía que se obtiene con la ingesta y la que se consume. El exceso de energía se almacena en forma de grasa corporal en los adipocitos que crecen y/o aumentan en número. Esta hipertrofia e hiperplasia de los adipocitos es la lesión patológica característica de la obesidad. El aumento en el tejido adiposo produce los problemas clínicos asociados con la obesidad, bien por el peso de la masa de la grasa extra o bien por el aumento de secreción de ácidos grasos libres, numerosos péptidos y otras adipoquinas por los adipocitos hipertrofiados. La obesidad y el sobrepeso asocian una serie de alteraciones endocrinas y metabólicas (tabla 1). La mayoría de los cambios se piensa que son secundarios a la obesidad, ya que se pueden inducir con la sobrealimentación y se eliminan con la pérdida de peso. La obesidad se acompaña de modificaciones en los niveles plasmáticos de determinadas hormonas y en cambios en sus patrones de secreción y/o aclaramiento. Algunas de estas alteraciones son secundarias a la obesidad mientras que otras podrían desempeñar un papel en su patogenia.
Principales cambios endocrinológicos en la obesidad
| GLÁNDULA ENDOCRINA | ALTERACIÓN HORMONAL |
| 1) Páncreas endocrino | Hiperinsulinemia |
| 2) Tejido adiposo | Hiperleptinemia |
| Descenso de adiponectina | |
| 3) Hipófisis | Disminución de GH basal y estimulada |
| Disminución de respuesta a estímulos de prolactina | |
| 4) Gónadas | Mujer: descenso de SHBG. Aumento de estradiol libre y testosterona libre |
| Hombre: descenso de SHBG. Descenso de testosterona total y libre | |
| 5) Suprarrenales | Aumento de cortisol libre urinario y cortisol plasmático normal |
| 6) Hormonas gastrointestinales | Descenso de ghrelina |
| 7) Tiroides | Aumento de TSH y T3 libre |
La alteración endocrinológica más característica de la obesidad es el incremento en la secreción de insulina. Las personas obesas tienen concentraciones de insulina aumentadas. La secreción de insulina basal e integrada de 24 horas es tres o cuatro veces mayor en sujetos obesos que en controles delgados6. Tanto la obesidad como la diabetes mellitus tipo 2 se asocian a resistencia a la insulina7, pero la mayoría de los sujetos obesos insulinoresistentes no desarrollan hiperglucemia. Para que la obesidad y la resistencia a la insulina se asocien con la diabetes tipo 2, la célula beta pancreática tiene que ser incapaz de compensar la disminución en la sensibilidad a la insulina7.
El tejido adiposo modula el metabolismo liberando ácidos grasos libres y glicerol, hormonas como leptina, adiponectina y citoquinas proinflamatorias como TNF alfa, o interleuquina 6. La liberación de ácidos grasos no esterificados es el factor aislado más importante que modula la sensibilidad a la insulina. El aumento de los niveles de ácidos grasos no esterificados (NEFA) se observan en la obesidad y la diabetes tipo 2, y se asocian con la resistencia a la insulina que vemos en ambas7.
La distribución de la grasa corporal es en sí misma un determinante de la sensibilidad a la insulina7. Individuos delgados con una distribución de la grasa periférica tienen mayor sensibilidad a la insulina que individuos delgados con una distribución de la grasa central. La grasa intraabdominal y subcutánea también son diferentes. La grasa intraabdominal es más lipolítica que la subcutánea y es menos sensible al efecto antilipolítico de la insulina8. Esta diferencia en las características de los adipocitos, junto con la proximidad al hígado de los depósitos de grasa intraabdominal hace probablemente que este órgano resulte más expuesto a los NEFA que los tejidos periféricos.
La sensibilidad a la insulina regula a la vez la función de la célula beta, que está casi siempre disminuida en la obesidad. Los NEFAS son importantes para el normal funcionamiento de la célula beta y potencian la liberación de insulina en respuesta a glucosa y a otros secretagogos. La elevación crónica de la glucosa plasmática y de los NEFA es deletérea para la célula beta, y se denomina glucolipotoxicidad7.
En resumen, en la obesidad es común el hiperinsulinismo, y la resistencia a la insulina es característica cuando existe una gran ganancia ponderal.
Tejido adiposoLa identificación y caracterización de la leptina en 1994 estableció firmemente al tejido adiposo como un órgano endocrino9. Se sabe que el tejido adiposo expresa y secreta una variedad de péptidos bioactivos, conocidos como adipoquinas, que actúan tanto a nivel local (autocrino/paracrino) como sistémico (endocrino). La importante función endocrina del tejido adiposo se enfatiza por los efectos adversos que tienen tanto el exceso como la deficiencia de tejido adiposo. El exceso u obesidad, especialmente en el compartimento visceral, se asocia con resistencia a la insulina, hiperglucemia, dislipemia, hipertensión arterial y estados protrombóticos y proinflamatorios. La prevalencia de obesidad y estas morbilidades asociadas, conocido como síndrome metabólico, ha alcanzado proporciones epidémicas. Es interesante que el déficit de tejido adiposo, conocido como lipodistrofia, también se asocie con características del síndrome metabólico tanto en humanos como en roedores. De las proteínas segregadas por el tejido adiposo, la mejor caracterizada es la leptina10,11.
Leptina (cuyo nombre deriva del griego leptos, que significa delgado) es un polipéptido de 16 kDa que contiene 167 aminoácidos con homología estructural a las citoquinas. En 1994 fue identificado el gen de la leptina9 y su receptor en 199512. Los adipocitos segregan leptina en proporción directa a la masa adiposa y al estado nutricional, y esta secreción es mayor desde la masa grasa subcutánea en relación con la de la masa grasa visceral. La secreción de leptina por gramo de tejido adiposo es dos veces mayor en obesos que en delgados.
Aunque inicialmente se veía como una hormona antiobesidad, el principal papel de la leptina es servir como señal metabólica de suficiencia energética más que de exceso. Los niveles de leptina descienden rápidamente con la restricción calórica y la pérdida de peso. Este descenso se asocia con respuestas fisiológicas de adaptación al hambre, que incluyen aumento del apetito y descenso del gasto energético. Estas mismas respuestas se observan en ratones y humanos leptin-deficientes a pesar de obesidad masiva. Además, estas respuestas se corrigen con la administración de dosis bajas de leptina. Por otro lado, las formas comunes de obesidad se caracterizan por niveles elevados de leptina circulante. Los mecanismos de resistencia a la leptina son desconocidos pero pueden resultar de defectos en la señal o el transporte de la leptina a través de la barrera hematoencefálica10. En humanos, las mutaciones de la leptina o de su receptor son extremadamente raras13.
Las personas obesas parece que son resistentes o insensibles a la leptina pues a pesar de la presencia de niveles elevados de leptina, que deberían reducir la ingesta de comida y grasa corporal, continúan manteniendo niveles elevados de grasa corporal.
Otras hormonas que son segregadas por el tejido adiposo con importantes efectos metabólicos son la adiponectina, la resistina, el TNFα, la interleuquina-6 (IL-6), proteínas del sistema renina-angiotensina, la adipsina, la proteína estimulante de la acilación (ASP) y la macrophages and monocyte chemoattractant protein-1 (MCP-1).
La adiponectina se segrega exclusivamente del tejido adiposo y es una proteína abundante en plasma. Con la excepción de casos de severa desnutrición y en el recién nacido, hay una fuerte correlación negativa entre las concentraciones plasmáticas de adiponectina en humanos y la masa grasa13. Los niveles de adiponectina están disminuidos en la obesidad mientras que la pérdida de peso los aumenta. Se ha visto que la adiponectina mejora la sensibilidad a la insulina en modelos de obesidad genética o inducida por la dieta13.
La hormona peptídica resistina (o FIZZ3) se identificó inicialmente como producida exclusivamente por el adipocito y se ha visto que juega un papel importante en la resistencia a la insulina inducida por la obesidad14. La resistina se expresa en los adipocitos de los roedores14 y en los macrófagos de los humanos. Sus niveles aumentan con la ingesta y en la obesidad y disminuyen con los ligandos PPARγ15.
La proteína enlazante del retinol-4 (RBP4) es una proteína de 21 kDa que fue descrita por primera vez como una adipoquina por Kahn et al16. RBP4 se ha visto que produce resistencia a la insulina alterando la señal de la insulina en el músculo16 y los adipocitos17.
HipófisisEn la obesidad, la alteración más clara en el sistema hipotálamo-hipofisario se relaciona con la hormona de crecimiento (GH). La secreción de hormona de crecimiento depende fundamentalmente de la interacción entre GHRH y somatostatina. Ghrelina, el ligando endógeno del receptor de los secretagogos de GH, probablemente desempeña también un papel18. Además, numerosos neurotransmisores, hormonas periféricas y señales metabólicas influyen en la secreción de GH19.
En la obesidad hay una disminución en la secreción de GH. Tanto en niños como en adultos, a mayor índice de masa corporal, menor respuesta secretora de GH ante diferentes estímulos secretores20, incluyendo la respuesta a GHRH21. Se ha visto que por cada unidad que aumente el IMC a una determinada edad se disminuye la secreción de GH hasta en un 6%. Este déficit relativo de GH en la obesidad puede contribuir al mantenimiento de la misma. Aparentemente todos los defectos en el eje GH-IGF1 en la obesidad son reversibles con la pérdida de peso, bien inducida por la dieta, bien a través de cirugía22. Cuando los sujetos obesos son tratados con GHRH y GHRP-6 a dosis altas, se produce un pico medio de respuesta de GH de 40μg/L, una respuesta masiva para los obesos20. El hecho de que las somatotropas, que han estado en un descanso funcional durante años o décadas de obesidad, respondan a esos estímulos combinados indica una función hormonal trófica hipotalámica normal y que las somatotropas no se vuelven atróficas en la obesidad20. La causa principal de la alteración en la secreción de GH en la obesidad puede ser una alteración en el hipotálamo, una función hipofisaria anormal o una perturbación en las señales periféricas que actúan a nivel hipofisario o hipotalámico.
En las personas obesas, la secreción de GH inducida por la administración exógena de ambos GHRH o GHRP-6 está bloqueada, descartando un déficit secretor de GHRH endógenos o del ligando natural del receptor de secretagogos de GH como factores causantes.
Los niveles elevados de insulina reducen la liberación de GH y la hiperinsulinemia que se encuentra con frecuencia en la obesidad se puede relacionar con esta alteración en la secreción de GH22,23.
La administración de una dosis baja de IGF-1 recombinante (rhIGF1) inhibe la respuesta de las somatotropas a GHRH tanto en obesos como en sujetos normales, indicando que la sensibilidad de las somatotropas al efecto inhibitorio del rhIGF1 se encuentra intacto en la obesidad, esto hace menos probable que la hiposecreción de GH de la obesidad sea debida a una inhibición de las somatotropas mayor de lo normal inducida por IGF-1 circulante.
Hay datos que sugieren que la disminución en la secreción de GH de la obesidad no se relaciona con los niveles elevados de leptina de la misma24.
La reducción de los ácidos grasos libres (FFA) con acipimox, un fármaco que disminuye los lípidos con mínimos efectos secundarios, aumenta de manera notable la secreción de GH inducida por piridostigmina, GHRH y GHRH + GHRP-6, restaurando el nivel de esta secreción a un 50-70% del normal25. Éstos y otros resultados nos indican que los niveles elevados de FFA juegan un papel importante en la disminución de GH en la obesidad y sugieren que el tratamiento con inhibidores de la lipolisis podría ser útil para restaurar la función somatotropa25. Por otro lado, la arginina y la piridostigmina, un fármaco que reduce el tono somatostatinérgico, aumentan la secreción de GH inducida por GHRH en sujetos obesos21. Esto unido a la secreción de GH casi normal en respuesta a hipoglucemia en obesos, sugiere que un aumento del tono somatostatinérgico puede parcialmente explicar la alteración en la función de las somatotropas en la obesidad. Aunque en la obesidad, el mecanismo responsable de la hiposecreción de GH sea probablemente multifactorial26, hemos visto que en sujetos obesos, la ghrelina es el estímulo secretorio de GH más potente hasta la fecha y que tras la administración combinada de ghrelina y GHRH se produce una secreción masiva de GH (fig. 1).
 media±EE tras la administración de GHRH, ghrelina (GHL) o GHL + GHRH en pacientes obesos.")
La persistencia de una respuesta más baja que en normales tras la administración de ghrelina solo o combinado con GHRH sugiere la existencia de otro defecto implicado en la alteración de la secreción de GH que existe en la obesidad. La secreción masiva de GH tras la administración de GHRH y ghrelina no se había observado con ningún estímulo previamente indicando claramente que la alteración en la secreción de GH en la obesidad es un estado funcional y potencialmente reversible y sugieren que la disminución en la secreción de ghrelina podría ser responsable al menos en parte de la hiposecreción de GH en la obesidad. Los niveles plasmáticos de ghrelina se encuentran disminuidos en los humanos obesos si se compara con los normales27.
Los secretagogos de GH (GHS) sintéticos se pueden considerar análogos de la ghrelina28 aunque todas las acciones conocidas hasta la fecha de estos compuestos no se pueden transferir de forma automática a la ghrelina. Nuestros resultados en obesos están de acuerdo con la evidencia de que los GHS producen una liberación de GH mayor que GHRH29. Nosotros hemos estudiado anteriormente la secreción de GH en obesos tras estímulo con el GHS sintético, GHRP-6, aunque existen importantes diferencias metodológicas entre ambos estudios, la respuesta de obesos a ghrelina fue mayor que la respuesta al secretagogo sintético GHRP6 tanto solo como en combinación con GHRH. Estos resultados sugieren que la ghrelina es un estímulo liberador de GH más potente que los GHS sintéticos. Como ya se suponía anteriormente, en base a los resultados de los GHS sintéticos20, los efectos aditivos entre la ghrelina y GHRH indican que dichos péptidos actúan, al menos parcialmente, por mecanismos de acción diferentes.
En resumen, en la obesidad existe una disminución en la secreción de hormona de crecimiento, la alteración de la función somatotropa en la obesidad es funcional y se puede revertir en diferentes situaciones. El mecanismo fisiopatológico responsable de esta hiposecreción de GH en la obesidad es probablemente multifactorial, la hipersecreción crónica de somatostatina, la elevación de los niveles plasmáticos de ácidos grasos libres y la disminución de ghrelina juegan un papel fundamental y, posiblemente, coexista alguna alteración menor en las células somatotropas (fig. 2).

Para el diagnóstico del déficit de GH del adulto se necesitan test de estímulo. La hipoglucemia insulínica (ITT) es el test de elección para el diagnóstico del GHD del adulto. La ITT es una prueba potencialmente peligrosa, además está contraindicada en muchas situaciones clínicas comunes en las que se puede sospechar un déficit de GH como puede ser cardiopatía isquémica, epilepsia y edad avanzada.
La obesidad es probablemente el mayor factor de confusión para el diagnóstico del déficit de GH del adulto, se sabe que la alteración en la secreción de GH en la obesidad es paralela a las alteraciones en la composición corporal como aumento de la grasa visceral, disminución de masa magra y de densidad mineral ósea. Hemos estudiado la respuesta de pacientes obesos normales y comparados con la respuesta de pacientes obesos con hipopituitarismo, ante diferentes estímulos30. La respuesta en ambos pacientes tras la administración de GHRH es similar. Sin embargo, la respuesta de GH tras la administración de GHRH y acipimox está marcadamente disminuida en adultos obesos con hipopituitarismo comparado con obesos normales. Con estos resultados se puede concluir que la secreción de GH tras GHRH y acipimox está disminuida en adultos obesos con hipopitiutarismo comparado con obesos normales, la prueba de GHRH y acipimox es segura y no tiene efectos secundarios por lo que se podría utilizar para el diagnóstico del déficit de GH del adulto21. Tras la administración de GHRH+GHRP-6 la respuesta máxima encontrada en pacientes con hipopituitarismo fue menor que la respuesta mínima en pacientes normales y en obesos. Sin embargo, tras ITT, GHRH o GHRH + acipimox la respuesta máxima en hipopituitarismo fue menor que la mínima respuesta en sujetos normales pero mayor que la mínima respuesta en obesos. Hemos encontrado también que el diferencial entre sujetos normales u obesos e hipopituitarios para GHRH + GHRP-6 fue mayor que para ITT, GHRH o GHRH+acipimox30 (fig. 3). Con estos estudios hemos demostrado que la administración de ambos acipimox o GHRP6 revierte el hiposomatotropismo funcional de la obesidad tras GHRH pero no son capaces de revertir el hiposomatotropismo orgánico del hipopituitarismo. La administración combinada de GHRH y GHRP6 es la prueba que mejor diferencia ambas situaciones, sin los efectos secundarios de la hipoglucemia insulínica30.
GónadasOvario (media ±EE) plasmática tras hipoglucemia insulínica (ITT), GHRH, GHRH+acipimox y GHRH+GHRP6 en pacientes con hipopituitarismo (, HIPO), pacientes obesos (¿) y controles normales (□).")
La obesidad en las mujeres se manifiesta como una serie de condiciones que preceden unas anomalías metabólicas como prediabetes, diabetes y enfermedad cardiovascular31. Éstas incluyen menarquia temprana, infertilidad y síndrome de ovario poliquístico (PCOS). Por otro lado, al final de la vida reproductiva de la mujer, el cese de la función ovárica, se asocia también con el desarrollo de obesidad, pues la menopausia precipita la ganancia de peso abdominal y se asocia con numerosas consecuencias metabólicas adversas.
En las mujeres, la obesidad se asocia con importantes alteraciones en los niveles de esteroides plasmáticos. La tasa de producción de testosterona, dihidrotestosterona y androstendiona se encuentra elevada en mujeres obesas mórbidas. La distribución corporal de la masa grasa es otro factor importante. La producción de hormonas sexuales y el metabolismo es diferente en obesas mórbidas con diferentes fenotipos. Mujeres con obesidad abdominal tienen una tasa de producción de andrógenos elevada y niveles mayores de testosterona libre y estradiol libre mientras que las mujeres con distribución glúteo-femoral de la grasa producen gran cantidad de estrona debido a la aromatización periférica de los estrógenos. Otros estudios han encontrado que en mujeres obesas premenopáusicas, un aumento de la grasa visceral se asociaba de manera significativa con niveles disminuidos de proteína enlazante de hormonas sexuales (SHBG) y el ratio 17 beta estradiol/testosterona libre y con niveles elevados de testosterona libre, después de ajustarlo por edad y por masa grasa total. En mujeres, la pérdida de grasa visceral se asocia con aumento en los niveles de SHBG y del ratio 17 beta estradiol/testosterona libre independientemente de la cantidad total de grasa perdida32. PCOS es la alteración ovárica más frecuente en mujeres premenopausicas y la obesidad es un hecho común en esta endocrinopatía. Dependiendo de la población estudiada, un 20-69% de las mujeres con PCOS son obesas (IMC>30) e independientemente de la obesidad, la mujeres con PCOS tienen un aumento de la acumulación de grasa a nivel intraabdominal.
Tras la menopausia, la dramática caída en los niveles de estrógenos junto con el relativo hiperandrogenismo contribuye a la ganancia de peso y al cambio en la distribución del tejido adiposo.
TestículoLa obesidad en los hombres se asocia con una reducción en la testosterona total y en los niveles de SHBG33,34. En el Massachusetts Male Ageing Study, los hombres que eran obesos al inicio y durante el seguimiento, bien medidos por IMC o bien por obesidad central (circunferencia de la cintura o índice cintura-cadera), tenían un mayor descenso en la testosterona total, libre y en la SHBG comparado con los hombres que nunca se habían clasificado como obesos35. Los factores patogénicos que se relacionan con esta reducción en los niveles de testosterona en la obesidad son la disminución de la capacidad de unión de la SHBG, la reducción en la amplitud de los pulsos de LH y la hiperestrogenemia.
La evaluación del perfil de hormonas sexuales en sujetos obesos muestra como los niveles de testosterona plasmática están disminuidos de manera moderada o masiva en hombres obesos a pesar de la reducción en las concentraciones de SHBG36. Se ha encontrado que el descenso de los andrógenos en los hombres obesos representa un continuo observable a cualquier grado de obesidad. Hay una relación inversamente proporcional entre la testosterona total plasmática, la testosterona libre y la SHBG con la masa grasa visceral. Isidori et al37 examinaron la relación entre las concentraciones de leptina y los niveles de hormonas sexuales en hombres adultos y encontraron que la leptina circulante se correlaciona con la testosterona total y libre incluso tras ajustarla con la SHBG, LH y estradiol y que la leptina era el mejor predictor hormonal de niveles de andrógenos disminuidos en hombres obesos.
La prevalencia del síndrome metabólico aumenta con la edad y está independientemente asociado a niveles menores de andrógenos y niveles mayores de estrógenos38. En modelos multivariable, la edad, la circunferencia de la cintura y el estado de salud se asocian con niveles de andrógenos bajos y con deficiencia de andrógenos sintomática, y de todas las variables, la circunferencia de la cintura es el factor asociado más importante39.
Glándula suprarrenalDada la relación entre obesidad, distribución de grasa central e hipercortisolemia (síndrome de Cushing), muchos estudios ha intentado determinar si el cortisol juega un papel en el desarrollo de obesidad en la población general, en la distribución central de la grasa en hombres y mujeres, y en los cambios en la composición corporal que se producen con la edad. En mujeres obesas con distribución abdominal de la grasa los niveles del cortisol libre urinario (por gramo de creatinina) se encuentran significativamente aumentados en comparación con las mujeres con una distribución periférica de la grasa o en controles normales. Los niveles basales de cortisol y ACTH aumentan significativamente tras la administración de CRF en todos los grupos pero este aumento es significativamente mayor en mujeres con obesidad abdominal que en mujeres con distribución periférica de la grasa o controles normales. Estos resultados, por tanto, sugieren que las mujeres obesas con distribución abdominal de la grasa tienen una hiperactividad del eje hipotálamo-hipófiso-adrenal (HHA)40.
Aunque de manera general se asume que los niveles plasmáticos de cortisol son normales en el síndrome metabólico y en la obesidad, existen respuestas dinámicas de cortisol exageradas41. En particular, la obesidad abdominal se asocia con un aumento en la excreción de cortisol libre urinario y aumento en las tasas de producción de cortisol total42. Estos datos sugieren que en la obesidad se producen pequeñas alteraciones en el mecanismo de retroalimentación, en el feedback inhibitorio del eje HHA y una alteración en el aclaramiento periférico a nivel hepático, con mínimas diferencias entre las manifestaciones del síndrome metabólico. Indicándonos que una producción excesiva de cortisol podría contribuir al desarrollo de la obesidad, sin embargo, no hay evidencia suficiente para afirmar de forma concluyente que un aumento en la secreción de cortisol es la causa del depósito excesivo de tejido adiposo en humanos. La secreción de cortisol en la obesidad se ha cuantificado por diferentes métodos, incluyendo dilución isotópica, medidas del cortisol libre en orina y los metabolitos C21. Estos estudios han demostrado de manera invariable aumento en la producción de cortisol en la obesidad43. Estudios recientes han encontrado aumentos similares en la producción de cortisol en obesas con síndrome de ovario poliquístico y obesas normales controles44.
La conversión local de cortisona a cortisol por la 11 beta hidroesteroide deshidrogenasa tipo 1 (11ß-HSD1) se ha relacionado de manera importante con la activación local del receptor de glucocorticoides, lo cual ofrece una explicación al paralelismo entre la obesidad idiopática y cushingoide. En humanos, la actividad del RNAm11ß-HSD1 está aumentada en el tejido adiposo subcutáneo abdominal de los sujetos obesos tanto in vivo como in vitro, en la mayoría de los estudios45. Hay estudios que sugieren que en algunos casos los niveles de 11ß-HSD1mRNA están también aumentados en el tejido adiposo omental visceral de mujeres obesas y es un fuerte predictor del tamaño de la célula adiposa en este depósito de grasa visceral46. En obesos con síndrome metabólico se ha descrito un aumento en la producción hepática de cortisol no acompañado de hipercortisolismo sistémico pues se compensa con un aumento del catabolismo hepático del cortisol y una menor actividad del eje corticosuprarrenal47.
Hormonas gastrointestinalesLa ghrelina es una hormona peptídica de 28 aminoácidos, producida predominantemente en el estómago y su estructura tiene una cadena de ácido graso en el tercer aminoácido del extremo N- terminal que es esencial para algunas de sus acciones biológicas, entre ellas su potente actividad estimuladora sobre la célula somatotropa. Estimula la secreción de hormona de crecimiento actuando directamente a nivel hipofisario a través del receptor de los secretagogos de GH20,29. Además de estimular la secreción de GH, ghrelina tienen otras acciones endocrinas y no endocrinas como estimular la secreción lactotropa y corticotropa, inhibe el eje gonadal, estimula el apetito y el balance energético positivo, controla la motilidad gástrica y la secreción ácida, actúa en la función pancreática tanto endocrina como exocrina, tiene acciones cardiovasculares, influye en el comportamiento y el sueño y modula la proliferación celular y del sistema inmune. Las células X/A de las glándulas oxínticas del estómago son la fuente principal de la ghrelina circulante. En el intestino delgado también se sintetiza ghrelina aunque en menor medida, siendo la cantidad de ghrelina producida menor a medida que aumenta la distancia del píloro.
La forma activa de ghrelina parece que necesita el ácido graso en el tercer aminoácido del extremo N terminal para sus acciones endocrinológicas, la forma no acilada no posee actividad endocrina28, sin embargo, no es biológicamente inactiva pues comparte con la forma acilada efectos antiproliferativos en líneas celulares cancerígenas de próstata, tiene efecto inotrópico negativo en el músculo papilar cardiaco y estimula la adipogénesis en la médula ósea, aunque los mecanismos de transducción de estos efectos precisan ser estudiados. Ghrelina parece tener un papel en la respuesta metabólica y neuroendocrina a la ingesta de comida. Sus niveles circulantes se encuentran elevados en la anorexia y la caquexia y disminuidos en la obesidad48,49 y los niveles plasmáticos de ghrelina se relacionan inversamente con el índice de masa corporal, la masa grasa corporal, los niveles de leptina plasmáticos y con los niveles de glucosa e insulina50.
Hay diferentes estudios que sugieren la importancia de ghrelina en la alimentación y el control del peso. El receptor de los secretagogos de GH (GHS-R1a) en el núcleo arcuato está implicado en la regulación de la secreción de GH, la ingesta de comida y la adiposidad51. Los niveles circulantes de ghrelina aumentan con el ayuno y descienden tras la ingesta de nutrientes tanto en ratones como en humanos52,53. Cummings et al53 demostraron picos preprandriales con supresión posprandial de los niveles de ghrelina en humanos al inicio de la ingesta voluntaria de alimentos. Estos datos sugieren que ghrelina juega un papel en el inicio de la ingesta53. Diversas evidencias sugieren un papel de ghrelina en la regulación a largo plazo del peso corporal y de la homeostasis energética. En humanos los niveles circulantes de ghrelina se correlacionan de manera inversa con el grado de adiposidad, con niveles menores en sujetos obesos y niveles elevados en condiciones como la anorexia nerviosa, enfermedades malignas o la caquexia asociada al fallo cardiaco crónico48,49. Los niveles de ghrelina aumentan tras la pérdida ponderal conseguida tanto con dieta solo como con una combinación de dieta y ejercicio y están suprimidos con la sobrealimentación y en el tratamiento de la anorexia nerviosa54. La ghrelina circulante en ayunas y tras la ingesta se encuentra alterada en la insuficiencia hepática y se recupera después del trasplante55,56. La neutralización de los niveles circulantes de ghrelina altera la recuperación del peso en el modelo animal57.
Los niveles plasmáticos circulantes de ghrelina aumentan antes de una comida y descienden tras la ingesta de nutrientes (fig. 4). La distensión gástrica que se produce con la infusión de agua en el estómago no produce reducción en los niveles de ghrelina, sin embargo, la ingesta de fibra no nutritiva sí que disminuye los niveles de ghrelina. Se ha demostrado que los sujetos obesos no presentan el descenso en los niveles plasmáticos de ghrelina que se observan tras la ingesta de comida en los delgados58. Aunque los obesos con síndrome de Prader-Willi, que se caracteriza por hiperfagia y obesidad, presentan niveles elevados de ghrelina, la concentración de ghrelina en ayuno en la mayoría de los sujetos obesos es menor que en los voluntarios con peso normal 59–61. La resistencia a la insulina se ha postulado como implicada en estos niveles menores de ghrelina en los obesos. El aumento en los niveles plasmáticos de ghrelina con la pérdida de peso inducida por la dieta es consistente con la hipótesis de que ghrelina juega un papel en la regulación del peso corporal a largo plazo en humanos. El bypass gástrico está asociado con niveles de ghrelina marcadamente suprimidos, lo cual probablemente contribuye al efecto reductor de peso de dicho procedimiento62. En resumen en la obesidad la ghrelina, la única hormona circulante orexigénica, está disminuida.

El péptido YY (PYY) es un péptido de 36 aminoácidos. PYY se sintetiza y se libera a la circulación por células enteroendocrinas especializadas denominadas células L que se localizan predominantemente en el tracto gastrointestinal distal. Se han descrito dos formas principales de PYY, PYY1-36 y PYY3-36. Los niveles circulantes de PYY aumentan en respuesta a la ingesta de nutrientes, con la carga calórica, la consistencia de la comida y la composición de los nutrientes afectando a sus niveles circulantes. El aumento inicial posprandial en los niveles circulantes de PYY se observa a los 15 minutos de la ingesta de comida, antes de que los nutrientes alcancen las células L del tracto gastrointestinal distal, por lo que hay mecanismos hormonales y neuronales implicados en esta liberación inicial. Los niveles de PYY típicamente alcanzan un pico 1-2 horas tras la ingesta, seguido por una fase de niveles estables de varias horas63. En 2002, Batterham et al describieron que la administración periférica de PYY3-36 reducía la ingesta de alimentos en roedores y en humanos con peso normal64. Estudios posteriores confirmaron que este efecto anoréctico del PYY3-36 estaba preservado en sujetos obesos. Además de contribuir a la saciedad posprandial, existen varias líneas de evidencia actuales que sugieren que PYY3-36 tiene un papel en la regulación del peso corporal a largo plazo65. PYY3-36 es selectivo para el receptor Y2 (Y2R). Un papel crucial de este receptor como mediador del efecto anoréxico del PYY3-36 durante la alimentación fue identificado inicialmente por Batterham et al quien encontró que los ratones sin el receptor Y2 eran resistentes a los efectos anorexígenos del PYY3-3664.
PP es un péptido de 36 aminoácidos producido predominantemente por las células F, localizadas en la periferia de los islotes de Langerhans del páncreas y en menor medida en el colon. Las concentraciones de PP circulante aumentan tras la ingesta de nutrientes de una manera bifásica y de manera proporcional a la carga calórica y sus concentraciones permanecen elevadas unas 6 horas tras la ingesta. Hay evidencias que sugieren que PP juega un papel en la regulación del peso corporal en roedores, pero el papel del PP en la patogénesis de la obesidad no sindrómica no está clara64.
El GLP1 (péptido similar al glucagón tipo 1) se produce en las células L del intestino delgado y se segrega como respuesta a los nutrientes, se conoce como una hormona incretina. Se libera a la circulación aproximadamente 30 minutos tras la ingesta de nutrientes. Su principal efecto es estimular la liberación de insulina dependiente de glucosa por los islotes pancreáticos. Reestablece la primera y segunda fase de secreción de insulina en respuesta a la glucosa66. Otras acciones que realiza son inhibir la liberación de glucagón tras la ingesta, enlentece el vaciamiento gástrico y reduce la ingesta de comida. El tratamiento con GLP-1 y sus análogos se asocia con pérdida de peso en parte debido a sus efectos en el vaciamiento gástrico y sus efectos secundarios gastrointestinales en forma de náuseas y vómitos.
Oxinomodulina (OXM) es una hormona peptídica de 37 aminoácidos aislada inicialmente de las células yeyuno-ileales porcinas. OXM se libera de las células L en respuesta a la ingesta de alimentos y proporcionalmente a la carga calórica. La administración central crónica de OXM, durante un periodo de siete días, reduce la ingesta de comida, la ganancia de peso y la adiposidad en ratas, sin evidencia de taquifilaxia. En humanos con peso corporal normal, OXM reduce la ingesta calórica inmediata en un 19%, la duración de este efecto inhibitorio tras la ingesta dura unas 12 horas.
Amilina es una hormona peptídica neuroendocrina de 37 aminoácidos. Se cosegrega con la insulina por las cèlulas beta pancreáticas en respuesta a la ingesta de nutrientes, a las hormonas incretinas y al impulso neural. Amilina posee acciones glucoregulatorias que complementan las acciones de la insulina, suprimen la secreción de glucagón posprandial y retrasa el vaciado gástrico.
TiroidesEl hipotiroidismo se asocial normalmente con una ganancia ponderal modesta, disminución de la termogénesis y del metabolismo, mientras que el hipertiroidismo se relaciona con pérdida de peso a pesar de aumento de apetito y aumento del metabolismo.
Aunque los obesos tienen habitualmente función tiroidea normal, se sabe que la TSH y el IMC se correlacionan de manera positiva. En efecto, en muchos estudios en niños, adolescentes y adultos han demostrado niveles de TSH discretamente aumentados en obesos comparados con sujetos normales delgados67. El aumento en los niveles de TSH no se relaciona con el déficit de yodo o con tiroiditis autoinmune en diversos estudios67. Mientras se ha encontrado que la obesidad aumenta la susceptibilidad a la enfermedad tiroidea autoinmune con un papel emergente de la leptina como determinante periférico68. Se ha sugerido una disfunción neuroendocrina que produce una secreción anormal de TSH como la causa de la elevación de las concentraciones de TSH en la obesidad, especialmente la leptina se ha visto que altera el eje hipotálamo-hipofisario69. Sin embargo, con el test de TRH se ha descartado una alteración en la respuesta de la hipófisis en niños obesos con niveles de TSH aumentados70. Por otro lado, el aumento en los niveles de TSH apunta hacia una resistencia a hormonas. El aumento de TSH y de hormonas tiroideas periféricas puede ser un proceso de adaptación al aumento del metabolismo energético basal y, por tanto, al gasto energético. En línea con esta teoría, los niveles elevados de TSH en la obesidad se normalizan tras una pérdida de peso sustancial67. El aumento moderado en los niveles de TSH de la obesidad se asocia con T4 normal, T4 libre normal y T3 libre y total aumentadas moderadamente, así como aumento en el volumen tiroideo67. Los niveles de T3 libre se encuentran aumentados en la obesidad debido a cambios en las vías de monodesyodinación. Posiblemente la elevación en las concentraciones de hormonas tiroideas en la obesidad aumenta el gasto energético y evita la acumulación de energía en forma de grasa. Como el ayuno y la pérdida de peso se asocian con una disminución en los niveles de hormonas tiroideas y por tanto un descenso en el gasto energético basal, este hecho probablemente contribuye a la dificultad que existe para mantener la pérdida de peso. Las vías fisiológicas que conectan la obesidad con el aumento de las hormonas tiroideas no están claras, la leptina puede ser uno de los vínculos entre el peso corporal y las hormonas tiroideas67. Se necesitan más estudios para determinar si una deficiencia leve de hormonas tiroideas y en consecuencia elevación de TSH por ejemplo al límite superior de la normalidad se relacionan con el desarrollo de la obesidad71. Por otro lado, aunque las hormonas tiroideas se ha usado de manera frecuente e inapropiada para conseguir perder peso en obesos eutiroideos, no hay indicación para su administración para controlar el peso corporal excepto en obesos hipotiroideos71.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los estudios referidos en este artículo han sido financiados por: FIS del Instituto de Salud Carlos III PI070413 y PI10/00088 y Xunta de Galicia PS07/12, INCITE08ENA916110ES, INCITE09E1R91634ES, IN845B-2010/187.
Agradecemos a Ramón Pensado su asistencia técnica.
