



Turner syndrome (TS) affects 1:2500 live females. It is caused by partial or complete absence of a sex chromosome. Patients with deletions of the distal segment of the short arm of X chromosome (Xp-) including haploinsufficiency of the SHOX (short stature homeobox) have, more often, short stature, skeletal abnormalities and hearing impairments. This article evaluates the current knowledge of the SHOX gene role in TS pathophysiology. Articles were searched from MEDLINE and LILACS databases, in the past 10 years, using the following keywords: Turner syndrome, SHOX gene, haploinsufficiency, short stature and hearing loss. As the inheritance of only one copy of the SHOX gene does not explain most of TS anomalies, more studies are needed to explain them. These studies will also improve understanding how SHOX participates in cartilage and bone growth and will help develop novel therapeutic strategies focused on SHOX-related disorders.
El sindrome de Turner (ST) afecta a 1:2500 mujeres vivas. Es causada por la ausencia parcial o completa de un cromosoma sexual. Las pacientes con deleciones en el segmento distal del brazo corto del cromosoma X (Xp-) que incluyen la haploinsuficiencia del gen SHOX (homeobox baja estatura), presentan más a menudo baja estatura, anomalías esqueléticas y problemas de audición. En este artículo se evalúa el conocimiento actual de la función del gen SHOX en la fisiopatología de ST. Se realizó una búsqueda de artículos científicos en MEDLINE y LILACS publicados en los últimos 10 años, mediante el uso de las siguientes palabras claves: síndrome de Turner, gen SHOX, haploinsuficiencia, baja estatura y pérdida de la audición. Como la herencia de una sola copia del gen SHOX no explica la mayor parte de las anomalías de ST, son necesarios nuevos estudios para explicar y mejorar la comprensión de cómo el SHOX participa en el crecimiento óseo y de cartílago. Estos nuevos estudios ayudarán a desarrollar nuevas estrategias terapéuticas dirigidas a los trastornos relacionados con el gen SHOX.
Turner syndrome (TS) is a genetic disease, caused by partial or complete absence of a sex chromosome, affecting 1:2500–1:3000 of live females.1,2 Classic clinical manifestations are short stature (final average height: 142–147cm); gonadal dysgenesis causing pubertal delay; primary amenorrhea; infertility and dimorphisms such as webbed neck and cubitus valgus.1–3
Besides the classic karyotype 45,X (present in 40–60% of the cases), other karyotype abnormalities can occur in TS, such as: duplications of the long arm (q) of the X chromosome with concurrent loss of short arm (p) to constitute a isochromosome (isoXq); ring formation (rX), deletions of the short and long arm of the X chromosome (Xp- ou Xq-), mosaicisms (45,X/46,XX) or karyotypes with the presence of the entire Y chromosome or parts of it.4 Although in 80% of the cases, the X chromosome is from maternal origin, it seems that imprinting is not a phenomenon in pathophysiology once the phenotype is not determined by the maternal or paternal origin of the X chromosome.5
Genetic anomaly is determined by the absence of genes on X chromosome. That is, the TS phenotype can be determined by a haploinsufficiency of genes bound to the X chromosome that escapes inactivation. Although one X chromosome undergoes inactivation in normal females during early embryogenesis, about 15% of all X chromosome genes, mostly situated on the short arm (Xp), remain active to some degree on both X chromosomes.6,7
Patients with TS and deletions at the end of the short arm of X-chromosome (Xp-), including haploinsufficiency of the SHOX gene (short stature homeobox), have short stature and orthopedic abnormalities (e.g.: cubitus valgus, Madelung deformity, micrognatia, and high-arched palate) and hearing impairment.8 Given the above, this article aims to present a review of current knowledge on the role of the SHOX gene in the pathophysiology of Turner syndrome.
SHOX geneHistoryThe SHOX gene (Short stature homeobox-containing gene) was first described, in 1997, as a gene responsible for the short stature in TS patients.9–11 Preliminary studies had showed that deletions at the end of the short arm of the X chromosome were almost always associated with short stature in TS patients.12,13 Thereafter, mutations in this gene were reported as responsible for short stature and orthopedic abnormalities in patients with Leri-Weill dyschondrosteosis and in patients with Langer mesomelic dysplasia.14–16 These mutations were also mentioned as the cause of the growth retardation in some individuals with idiopathic short stature (ISS).17
EpidemiologyThe global estimation of the deficiency incidence of the SHOX gene is between 1:2000 and 1:5000 in general population and 1:40 and 1:150 among individuals with short stature.18,19
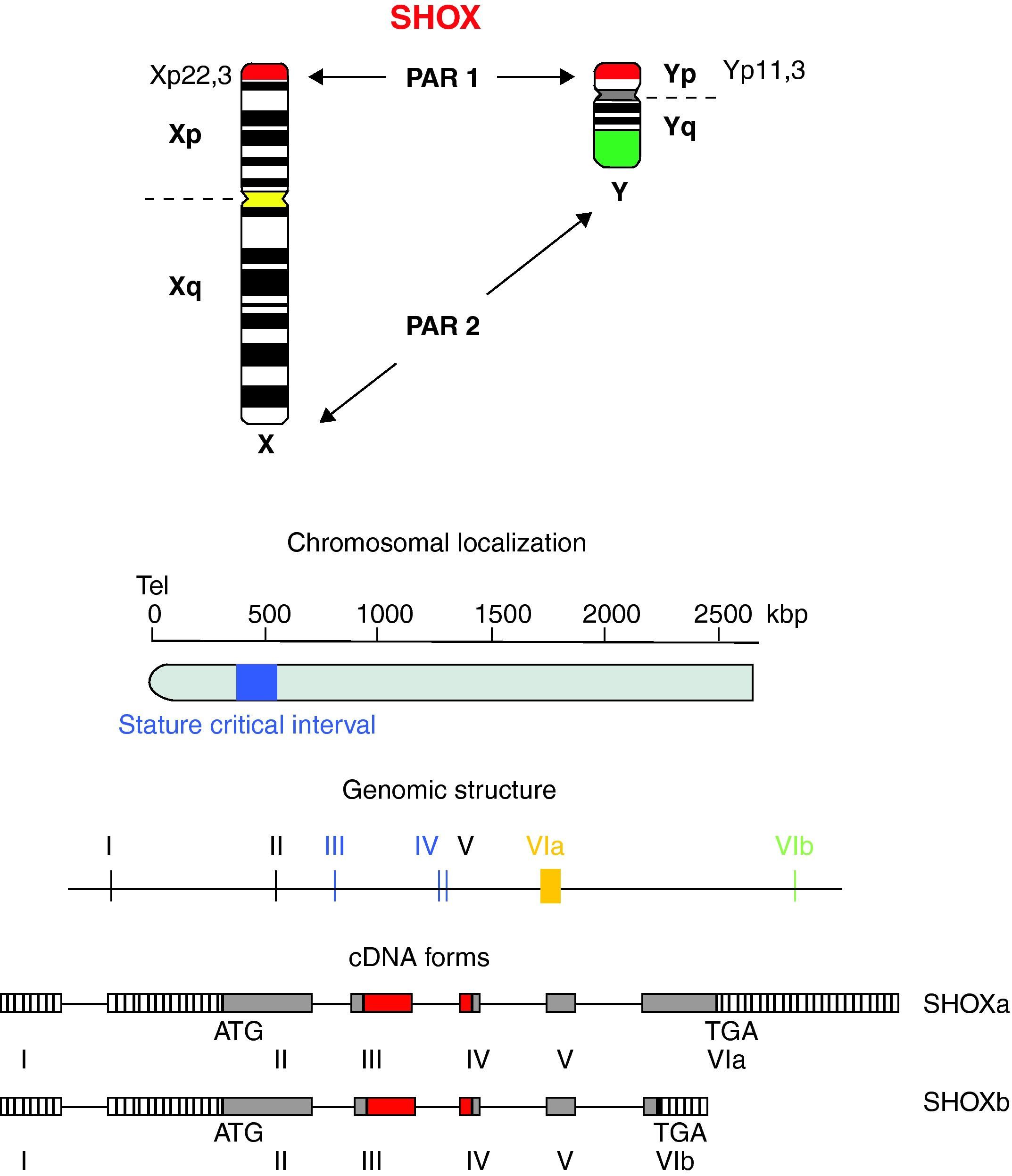
Location and genomic structureHomeobox is a 180-bp DNA sequence that codes for a 60-amino acid DNA-binding gene region called homeodomain.8,18,19 The SHOX gene is located in the pseudoautosomal region 1 (PAR1), a DNA segment with about 170-kb, 500kb distant from telomeres and situated at the end of the short arm of the X and Y chromosomes (Xp22.3 and Yp11.3) (Fig. 1).5,20
. SHOX: short stature homeobox.")
Although one X chromosome undergoes inactivation in normal women, several genes of the “inactive” X escape from inactivation. The SHOX gene is one of these, being inherited, as two functional copies, from both parents.8,18 The loss of one of these non-inactivated genes may cause some of the anomalies in TS and other syndromes.
The SHOX gene consists of 7 exons, being 1 non-coding and 6 coding exons.8,18 The homeobox domain is codified by the exons 3 and 4 that act as transcriptional activators. Other homeodomain, known as OAR, situated at the C-terminal end, is fundamental to keep genetic transactivation capacity.
The SHOX gene also encodes two alternatively spliced transcripts named SHOXa and SHOXb. Both transcripts are identical at the 5′ end but differ at the 3′ end in the last exon and are translated into two protein isoforms of 292 (SHOXa) and 225 (SHOXb) amino acids, the latter lacks the C-terminal portion which harbors a 14 amino acid motif known as OAR domain (otp, aristaless, and rax) that was thought to be important for transcription activity and be involved in protein–protein interactions or DNA binding.21,22 Nucleotide sequence analysis suggests that the human and mouse SHOX2 shares 99% identity at the amino acid level and generates two isoforms by alternative splicing, SHOX2a and a shorter version SHOX2b. Both isoforms contain the sequence encoding the homeodomain, an SH3 binding domain, and the OAR-domain. However, SHOX2b lacks 363 nucleotides corresponding to the N-terminus of SHOX2a and 36 nucleotides corresponding to the C-terminus of SHOX2a.21
Having assigned the SHOX transactivating activity to the most C-terminal part of the protein is interesting because any nonsense mutation leading to a SHOX truncation upstream of the OAR domain would lead to a short stature or LWS phenotype. Besides that, SHOXb is predicted to be inactive as a transcriptional activator. Consequently, SHOXa/SHOXb heterodimers and SHOXb homodimers are predicted to have transcriptional activation activities different from those of SHOXa homodimers. Therefore, SHOXb represents an excellent candidate for a modulator of SHOXa activity. The idea of such modulating activities of the SHOXb isoform is in agreement with reports on other developmentally important transcription factors. Additional attractive candidates for SHOX modulators might be encoded by the SHOX2 gene. Given an identical homeodomain of SHOX and SHOX2, these proteins may compete for identical binding sites or even directly interact with each other.23
ExpressionThe SHOX gene codifies a homeodomain transcription factor expressed during fetal life in the development of bone tissue in the distal humerus, radius, ulna, wrist, similar bones in the legs, first and second pharyngeal arches. The protein is specifically expressed in the growth plate of hypertrophic chondrocytes and seems to have a main role in the regulation of the differentiation and proliferation of these cells.24 During the embryonary period, the expression of the SHOX is restricted to limbs and pharyngeal arches and can be detected in the osteoblasts from the second month of gestational age on.10
Within the limb, SHOX is initially expressed in undifferentiated mesenchymal tissue.11,25 When the mesenchyme condenses, and chondrification takes place, SHOX is expressed most strongly in perichondrial layer. In this layer, chondroblasts (and subsequently osteoblasts) differentiate, prior to laying an envelope of cartilage surrounding the mesenchyme condensation. This specific expression in developing bone suggests that SHOX is involved in bone morphology. Besides that, SHOX2 is expressed more proximally and medially, and is primarily in connective and muscular tissues. In pharyngeal arches, SHOX is expressed primarily in the mesodermal core, whereas SHOX2 expression is more intense in the outer mesenchymal layers.25
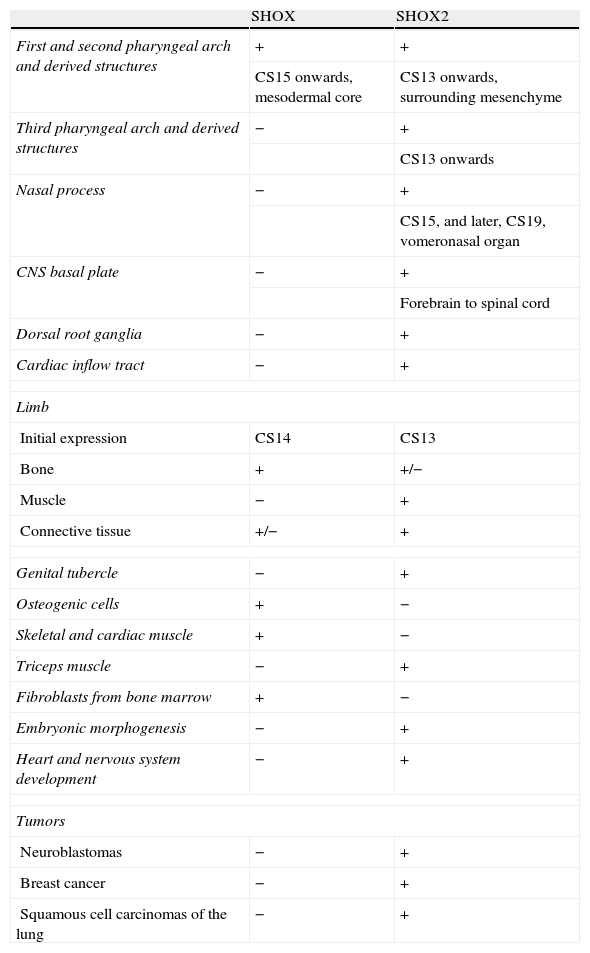
The SHOX gene is highly expressed in osteogenic cells, but its expression was also described in skeletal and cardiac muscles and in the fibroblasts from the bone marrow. During the embryonic period, its expression is restricted to members and pharyngeal arches, and it can be detected in osteoblasts of human embryos from the second month of pregnancy on.10 Therefore, SHOX expression is not only restricted to the developing limbs but was also detected in the mesenchymal core of the first and second pharyngeal arches. Expression was first seen at Carnegie stage (CS) 15. The first pharyngeal arch mesenchyme goes on to produce the maxilla and mandible as well as some of the bony elements of the external and middle ear. The mesenchyme of the second branchial arch contributes to several bones, including those of the middle ear.25 These findings support the idea that the gene is essential for bone development.10 A summary of SHOX and SHOX2 expression is given in Table 1.
The expression of SHOX and SHOX2.
| SHOX | SHOX2 | |
| First and second pharyngeal arch and derived structures | + | + |
| CS15 onwards, mesodermal core | CS13 onwards, surrounding mesenchyme | |
| Third pharyngeal arch and derived structures | − | + |
| CS13 onwards | ||
| Nasal process | − | + |
| CS15, and later, CS19, vomeronasal organ | ||
| CNS basal plate | − | + |
| Forebrain to spinal cord | ||
| Dorsal root ganglia | − | + |
| Cardiac inflow tract | − | + |
| Limb | ||
| Initial expression | CS14 | CS13 |
| Bone | + | +/− |
| Muscle | − | + |
| Connective tissue | +/− | + |
| Genital tubercle | − | + |
| Osteogenic cells | + | − |
| Skeletal and cardiac muscle | + | − |
| Triceps muscle | − | + |
| Fibroblasts from bone marrow | + | − |
| Embryonic morphogenesis | − | + |
| Heart and nervous system development | − | + |
| Tumors | ||
| Neuroblastomas | − | + |
| Breast cancer | − | + |
| Squamous cell carcinomas of the lung | − | + |
SHOX: short stature homeobox
The SHOX gene is not expressed in the vertebrae, phalanges, heart, central nervous system or genitalia.11,24,26,27 On the other hand, the observed stature changes and dysmorphic skeletal features in TS imply that the SHOX gene influences the timing of growth plate fusion and skeletal maturation. SHOX gene is expressed in the middle aspect (mesomelic region) of the upper limbs and the pharyngeal arch and has undetectable expression in embryonic somites that eventually form vertebrae.11
Expression of homeobox proteins themselves is controlled both on the transcriptional and translational levels. SHOX2 is a known regulator of chondrocyte hypertrophy and has important functions in skeleton development and embryogenic pattern formation. Other regulatory functions affect embryonic morphogenesis, heart and nervous system development.28SHOX2 is expressed in embryonic and adult skeletal muscles of the proximal limb.29
Interestingly, SHOX2 expression is frequent in various different types of tumors, among them neuroblastomas, breast cancer and squamous cell carcinomas of the lung. Homeoproteins are often found to be deregulated in cancer and both down- and up-regulation can be linked with tumor development and progression by activating or repressing multiple downstream genes, thereby acting as protooncogenes or tumor suppressor genes. However, a direct or indirect implication of SHOX2 as transcriptional regulator during cancerogenesis can be hypothesized.28
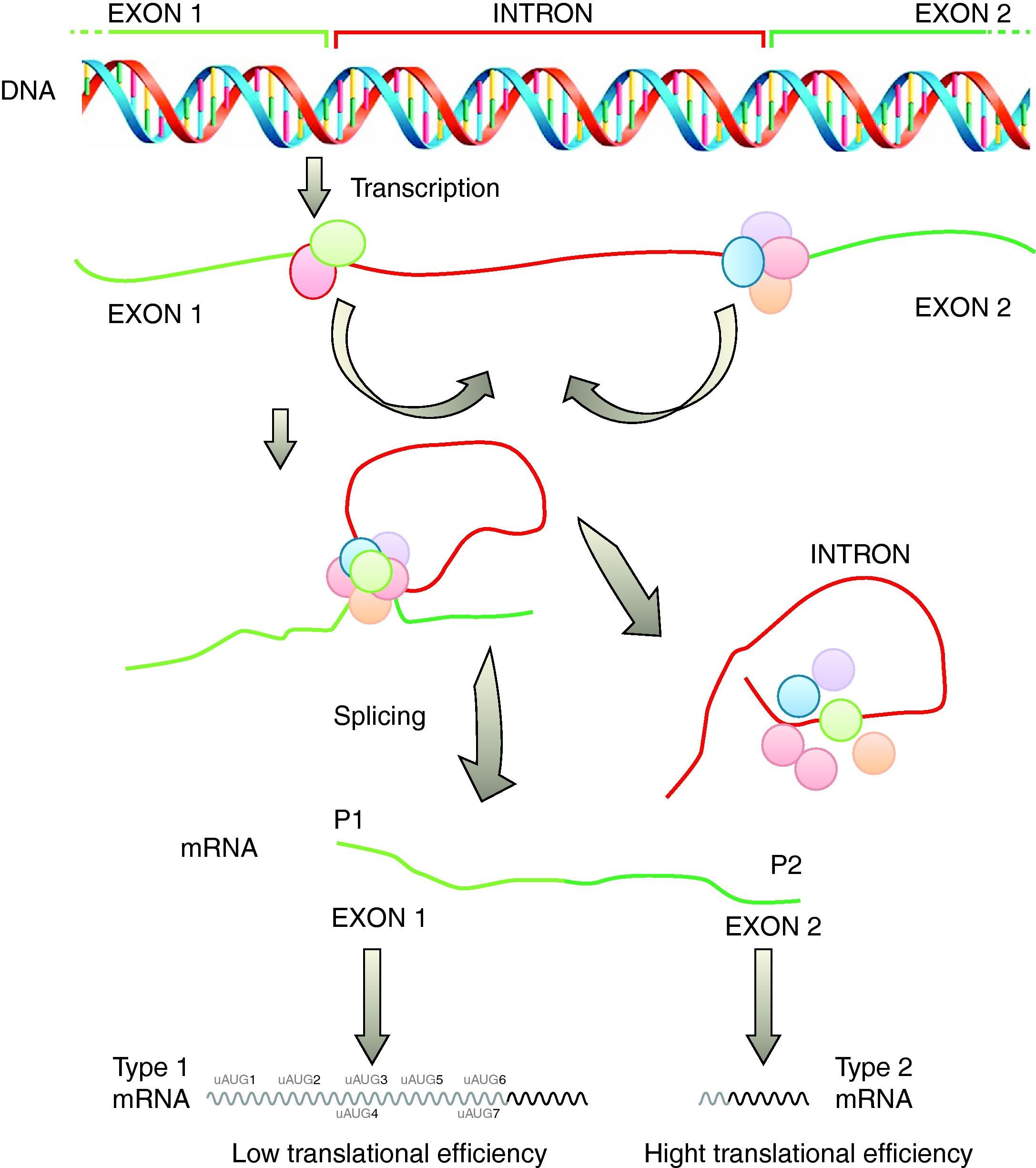
TranscriptionAlthough initially transcriptional regulation has been correlated with a variety of human diseases, it has become increasingly clear that post-transcriptional processing and differential translation represent equally important checkpoints in the tissue-specific and disease-related control of gene expression. An example of such importance is the investigation of structural and functional properties of the 5′-UTR of the SHOX mRNA by computational, in vitro, cell culture based and in vivo analyses revealed some interesting characteristics. In vitro and cell culture studies demonstrated an alternative intragenic promoter within exon 2 of the SHOX gene.30
Another important checkpoint is related to the results reported in several studies which were integrated into a combinatorial model of SHOX gene expression as depicted in Fig. 2. According to this model, SHOX expression is regulated on the transcriptional level by alternative promoters generating type 1 or type 2 transcripts with identical coding capacities but distinct translation efficiencies. This unique situation suggests that intragenic promoter (P2) is utilized in situations with immediate needs of high SHOX amounts, whereas upstream promoter (P1) allows the generation of transcripts that facilitate fine tuning of protein levels by translational control mechanisms possibly related to cellular stress situations.30
. SHOX: short stature homeobox.")
Transcription of the SHOX gene is regulated by alternative promoters generating different type 1 or type 2 transcripts with identical coding capacity but distinct 5′-UTRs. These 5′-UTRs exhibit significant differences in their translation activity due to the presence of seven uAUGs in type 1 transcripts (adapted from Blaschke et al.30). SHOX: short stature homeobox.
On the other hand, a major role in the regulation of human height has been attributed to the transcription factor and homeodomain protein SHOX. To address which genes are influenced and controlled by the expression of SHOX, the transcriptional profiling was carried out. The gene with the most significant and by far strongest upregulation upon SHOX induction was the NPPB gene encoding the peptide brain natriuretic peptide (BNP).31 Besides that, it is known that two SHOX isoforms exist, SHOXa and SHOXb, both contain a homeodomain, but only SHOXa (SHOX) acts as a transcriptional activator in osteogenic cells via its transactivation domain, the OAR (otp, aristaless and rax), which is absent in SHOXb. Recently, natriuretic peptide B (NPPB), which encodes brain natriuretic peptide, an important regulator of endochondral ossification, has been identified as the first transcriptional target of SHOX.20
Relationship of SHOX gene and fibroblast growth receptorSHOX is able to regulate the expression of fibroblast growth factor receptor gene (FGFR3) in different cell types and in chicken micromass (chMM) cultures. On the other hand, defined FGFR3 upstream region has a different impact in SHOX-dependent transcriptional regulation in normal human dermal fibroblast (NHDF) and U2OS (human osteosarcoma cell), suggesting the presence of individual regulatory regions with different responses to cell-specific cofactors. Thus, it is possible to claim that, depending on the cellular environment, the availability of cofactors and/or the specific binding site, SHOX can act as either an activator or a repressor on the expression of FGFR3.32
It was shown in several in vitro and in vivo systems that SHOX represents a strong regulator of FGFR3. A positive regulation of FGFR3 by SHOX, as detected in U2OS and NHDF cells, may not be compatible with respect to the clinical manifestations in the limbs of affected patients. In contrast, a repressive effect of SHOX on FGFR3 expression as observed in chMM cultures, which represents a more suitable model system for limb development, can much better explain the underlying mechanisms of the human phenotypes. A negative regulation is also consistent with the mutually exclusive expression patterns of SHOX and FGFR3 detected in developing chicken limbs. The observed FGFR3 repression might be particularly relevant in mesomelic segments where SHOX is predominantly active.32
In healthy individuals, SHOX dependent repression would generally result in a lower expression of FGFR3 in the mesomelic segments compared with the rhizomelic segments where SHOX expression is much weaker. Consequently, SHOX deficiency would create a relatively increased FGFR3 expression in ulna and radius as well as in tibia and fibula, thereby accelerating a fusion of the growth plates and causing a relative shortening of the respective bones. The effect on the rhizomelic segments should be less pronounced, which is consistent with the human phenotypes. FGFR3 gain-of-function mutations, on the other hand, would have a stronger effect on the upper arm and leg, since FGFR3 is not targeted by SHOX in that part of the limbs and therefore expressed at higher levels than in the mesomelic segments. This overrepresentation of FGFR3 would result in an earlier growth plate closure of humerus and femur compared with the lower limb segments, which is in line with the rhizomelic short stature found in achondroplasia patients.32
MutationsThe high frequency of repetitions in the PAR1 region makes this genomic region more prone to recombination and that is why the deletions are found in 70% of the affected individuals.15,18,33 Most of the mutations were described in patients with Leri-Weill dyschondrosteosis. These mutations are present all over the codifying region, including substitutions, deletions and insertions.18 Most mutant alleles are inherited.18,34 The loss of one of the copies of the SHOX gene, a mandatory heterozygous deletion, is responsible for 2/3 of the short stature of TS patients and the loss of two copies causes Langer mesomelic dysplasia.10,35
SHOX gene function is dosage dependent: the loss-of-function mutation of one SHOX allele (haploinsufficiency) results in the disorder of SHOX deficiency which causes growth failure. The most frequent SHOX mutations found are gene deletions of different size which encompass the SHOX gene itself or a regulatory enhancer region which is located 50–250kb downstream of the coding region. These deletions account for around 80% of all mutations. The other gene defects found are missense and nonsense mutations which are spread all over the gene, but most of them are located within exons 3 and 4 which encode the functional important homeodomain. These mutations are predicted to cause inactivation of the protein or to block nuclear translocation or dimerization of SHOX.36
Turner syndrome is almost always associated with the loss of one SHOX gene because of the numerical or structural aberration of the X chromosome associated with this syndrome. The loss of both SHOX alleles causes the complete lack of SHOX and an extreme phenotype of osteodysplasia called Langer syndrome. The gain of 1 or 2 additional copies of SHOX due to structural aberrations of the X chromosome can be associated with tall stature. On the other hand, the prevalence of SHOX mutations in children with so-far-unexplained short stature (idiopathic short stature) has been estimated according to SHOX mutation screening studies. Estimates for prevalence in short children ranked from 2 to 15%. The prevalence of SHOX mutations in individuals with Leri-Weill syndrome is around 50–90%.36
In contrast to many other growth disorders like growth hormone deficiency or even idiopathic short stature, in SHOX deficiency females outnumber males. This phenomenon has been explained by the more severe phenotype in females which may cause ascertainment bias. Another explanation for the female preponderance in SHOX deficiency may be a genetic one: SHOX (X) deletions have been more frequently reported than SHOX (Y) deletions, which may indicate that the SHOX on the X is more prone to getting deleted than the SHOX on the Y.36
In SHOX deficiency, short stature is accompanied and to an important extent caused by (mesomelic) shortening of the extremities. Therefore, clinical judgment requires measurement of the arm span and sitting height as well as calculation of the subischial leg length besides measurement of the standing height. A helpful parameter which integrates these 3 parameters is the extremities trunk ratio. The ratio has the sum of the leg length and arm span in its numerator and the height of the trunk in its denominator. In children with SHOX deficiency, this ratio is significantly lower than in controls. A normal ratio in a school child excludes SHOX deficiency with a high negative predictive value at almost 100%.36
Genetic testingLarge deletions, such as those that occur in total or partial monosomies of the X chromosome of TS can be diagnosed by the lymphocyte karyotype in the peripheral blood.10 If the karyotype does not allow the diagnosis, one should order other tests to detect this abnormality. There is no consensus of which test should be requested next. Depending on laboratory availability, fluorescence in situ hybridization (FISH) could be the initial test. If no deletion is detected, sequence analysis or mutation scanning can be ordered. More recently, Funari et al.14 studied three methodologies to detect SHOX deletions: fluorescence in situ hybridization (FISH), microsatellite analysis and multiplex ligation-dependent probe amplification (MLPA), concluding that the MPLA was more sensitive and less expensive and should be used as the initial molecular test to detect SHOX gene deletions. D’haene et al.37 has indicated a quantitative polymerase chain reaction (qPCR) as a reliable, accurate and cost efficient test to screen the SHOX gene region.
SHOX gene and the pathophysiology of Turner syndromeMost TS individuals have only two copies of the SHOX gene. Nevertheless, the haploinsufficiency of the SHOX gene does not explain all the anomalies in TS, suggesting that other genes take part in this process. Table 2, modified from Hjerrild et al., shows the dimorphisms associated with the SHOX gene.4
Haploinsufficiency of the SHOX gene and phenotypical manifestations of Turner syndrome (modified from Hjerrild et al.4).
| Clinical findings probably explained by haploinsufficiency of theSHOXgene |
| - Short stature (responsible for the majority of the height deficit) |
| - Skeletal malformations: short 4th metacarpals and metatarsals, cubitus valgus, mesomelia (shortening of the forearms and lower legs), Madelung deformity, high-arched palate, micrognathia |
| - Sensorineural hearing loss |
| Clinical findings probably not explained by haploinsufficiency of theSHOXgene |
| - Congenital heart disease |
| - Renal malformations |
| - Ovarian insufficiency |
| - Endocrine disorders |
SHOX: short stature homeobox
The linear growth of the TS is defective since intrauterine life and tends to decelerate by 5–7 years-old.38 The pubertal spurt is absent even in those girls with spontaneous pubertal development. These alterations make short stature (a 20cm deficit in the final height) one of the main features of Turner syndrome.24
The discovery of the SHOX gene in the pseudoautosomal region 1 (PAR1) of the sexual chromosomes, along with its role in bone growth process, collaborated in the understanding of short stature in Turner syndrome.17,38 Most women with TS inherit just one copy of the SHOX gene. This state of haploinsufficiency seems to be substantially responsible for the height deficit in these patients.39 Other evidence for SHOX as the gene determinant of short stature in TS, comes from XY girls with interstitial Yp deletions. As these patients present several TS stigmata, although they have a normal height, this suggests that the growth gene of Turner dwells both in the Yq (and its respective partner in X chromosome) and inside the pseudoautosomal region of the sexual chromosomes, that is present in a triple dosage in these women.40
The deficiency of the SHOX gene seems to be one of the main causes of short stature in patients with Leri-Weill syndrome (LWS), Langer mesomelic dysplasia and also of proportional short stature in patients with isolated short stature.19,41 More recently, several studies consolidated the defects in the SHOX gene as the main monogenic causes of short stature.10 On the other hand, trisomy of the SHOX gene in Klinefelter syndrome or Triple X patients, causes tall stature.19
Nevertheless, these isolated defects in the SHOX gene are unlikely to explain fully the growth abnormalities in TS.27 Therefore, variations in the inactivation of this gene lead to several levels of haploinsufficiency and can explain, partially, the broad phenotypical variance. Additional factors such as genes that determine height, aneuploidy, chromosome unbalance, ovarian failure and delayed puberty can contribute.24
Skeletal abnormalitiesThe SHOX gene is expressed neither in the axial skeleton development nor in the skull. In other affected regions, skeletal defects are present in a large phenotypic variation, according to the severity of expression and bone involvement, and tends to worsen with puberty.42,43 For example, mutations and deletions can be found in individuals with short stature without any skeletal abnormality.43
The main skeletal abnormalities in TS, related to SHOX deletions are micrognatia, cubitus valgus, high-arched palate and short metacarpals and metatarsals.1,19,44 Only 8% have Madelung deformity which is characterized by shortening and bowing of the radius and dorsal dislocation of the distal ulna.45–47 It is possible to infer, therefore, that patients with short stature, normal karyotype and presence of one or several skeletal abnormalities associated with TS can be carriers of the SHOX gene.19,44,48
Because reduced expression of SHOX is associated with skeletal growth plate dysmorphism in TS, there is reason to postulate that the same gene abnormality is associated with TS scoliosis (and kyphosis).11 Typical idiopathic-type scoliosis develops in up to 10% of TS girls, most commonly during adolescence. Progression of preexisting scoliosis has been reported to be a possible side effect of GH therapy, albeit the association is not fully established.49 To date, no data regarding SHOX gene expression in vertebral growth plates in TS or other conditions are known.11
The skeletal radiological abnormalities in patients with SHOX defects include: coarse trabecular pattern, short metacarpals and metatarsals, radial and tibial bowing, abnormal tuberosity of humerus, abnormal femoral neck, triangulation of the radial epiphyses and exostoses of the proximal tibia and fibula.8,50
Non-skeletal abnormalitiesUp to now, SHOX expression was not detected in cardiac, renal or vascular organogenesis. This fact suggests that SHOX probably does not have a role in the development of non-skeletal somatic features in the Turner phenotype.25 The haploinsufficiency of the SHOX does not contribute to other clinical manifestations of Turner syndrome, such as ovarian failure and lymphedema, either.23,51
Ovarian failure and defective production of estrogens are present in most patients due to precocious follicular apoptosis. Some girls can present spontaneous pubertal development and even regular menses52. Curiously, the spontaneous production of estrogen and its replacement seem to enhance the defect of the gene SHOX, causing a faster advance of the bone age with precocious closing of the epiphyseal plate.42,44
Growth hormone treatmentPatients with LWD, or ISS, which show haploinsufficiency of the SHOX gene by point mutation or deletion, present short stature similar to that observed in girls with TS and, by analogy, it can be assumed that they would present, in a similar manner, the benefits of use of recombinant human growth hormone (hrGH). A recent study showed that patients with haploinsufficiency of SHOX show growth response to treatment with hrGH similar to patients with ST and with significant gain in height compared to untreated patients.10 The prepubertal patients with haploinsufficiency of the SHOX gene and treated with hrGH with the dose of 50μg/(kgday) (equivalent to 0.15U/(kgday)) showed increased growth velocity (GV) (4.8±0.3cm/year before treatment, 8.7±0.3cm/year in the first year and 7.3±0.2cm/year in the second year of treatment), while untreated patients remained stable in both the GV years of observation.10 This increase in GV was reflected in an improvement in the Z height of −3.3±0.2 pretreatment to −2.1±0.2 after two years of treatment and average gain of 5.9cm more than the control group in the same period. Patients with phenotype of DLW and EIB had a similar response to treatment with hrGH. However, there are no data on final height in children with SHOX defects treated with hrGH.10
Short stature is the most consistent feature in TS patients and many treatments have been used to improve final height outcome in these patients. Treatment with GH improves growth velocity and final height in most of these patients. When GH therapy is initiated at 9 years of age, final height improves by approximately 8cm, against a height gain of approximately 6cm when therapy is started at 11–13 years. Recommendations for the diagnosis and management of TS state that “..initiation of GH therapy should be considered as soon as a patient with TS has dropped below the 5th percentile of the normal female growth curve”.49 A study involving a nationwide growth hormone (GH) treatment program for Turner syndrome (TS), focusing on the first-, second-, and third-year responses of all prepubertal girls receiving GH for TS in Australia in 2007, showed some interesting results.53 The first year of treatment is particularly important as response is greatest and is most amenable to manipulation by dose variation. The effect of a dose increment after the first year cannot reverse the normal decline in response. It is also when dose and age at commencement interact, with younger patients responding disproportionately better to higher doses. These factors, but in particular first-year dose, may also set the potential for response, possibly over the whole period of treatment. The analysis supports initiating GH treatment of Turner syndrome as early as possible with the highest safe dose, at least 9–5mg/(m2week) (0–39mg/(kgweek)), in the first year.53
Hearing impairmentThe diseases of the middle ear start in childhood, being responsible for a morbidity parcel of these patients.5 Recurrent suppurative otitis media and cholesteatoma are the most common disorders. The hearing loss can be conductive, sensorineural or mixed, whose prevalence increases with age.54
Patients with SHOX gene deficiency have a higher propensity to middle ear infections due to skeletal abnormalities of the outer ear.25 It is suggested that the high occurrence of otitis media is due to the reduced size of the mastoid and the skull base, which cause tube dysfunction and decreases the middle ear ventilation.55 All the structures involved in these dysfunctions (mandible, auditory ossicles and outer ear) are composed from the first and second pharyngeal arches mesenchyma, where the SHOX gene is expressed. The more substantial the loss of genes in the short arm (p) of the X chromosome, where the SHOX gene is, the worse will be the growth retardation of the temporal and auricular bones.25,55
The dose–response relationship between hearing and amount of genes on the p-arm of the X chromosome supports the suggestion of a cochlear growth disturbance caused by the lack of growth-regulating genes such as the SHOX gene. The organ of Corti may possibly be shorter and/or the number of sensory hair cells lower in TS. If the amount of growth-regulating genes determines the total number of sensory hair cells, the fewer sensory hair cells at birth, the more rapidly will the age-related degeneration of the outer hair cells affect the hearing function. The sensorineural hearing loss and especially the age-dependent high frequency hearing may be a result of a reduced number of sensory hair cells already at birth.55
Conclusions and perspectivesThe SHOX gene is related to a peculiar situation, where mutations affecting a single gene can cause multiple phenotypes. Thus, more studies are needed in order to increase the understanding of the function of this gene. Among these researches, the following perspectives emerge: (I) As the first year of rhGH (recombinant human growth hormone) treatment, used in children with TS, had the greatest response and is most amenable to manipulation by dose variation, it is feasible that the same therapy can improve the growth rate and final height of children with SHOX mutation without TS; (II) Other interventional approach for short stature treatment could be the use of aGnRH (gonadotrophins releasing hormone analog), which suppress the production of gonadal steroids and can attenuate the development of bone alterations, increasing the growth period; (III) As there is no correlation between the type and position of the gene mutation and the subsequent phenotypes, the elucidation of the signaling cascades that are activated by the transcription factor SHOX inside the growth plate could lead to a better understanding of the complex process involving bone formation; (IV) Since the SHOX gene seems to be related with several clinical conditions, previously considered as distinct clinical syndromes, it is possible that its haploinsufficiency can also mediate other mild skeletal dysplasias.
Conflict of interestThe authors declare that there is no conflict of interest.
