


El ganglioneuroma (Gn) es un tumor benigno del sistema nervioso simpático que puede aparecer a lo largo de la cadena simpática ganglionar paravertebral, desde el cuello hasta la pelvis, y en ocasiones en la médula adrenal. El Gn suprarrenal (GnS) suele presentarse en menores de 20 años de forma asintomática y se diagnostica incidentalmente en pruebas de imagen solicitadas por otro motivo. A pesar de su benignidad, hay casos documentados de malignización a schwanoma maligno y se ha asociado a tumores como el feocromocitoma. Su tratamiento es la exéresis quirúrgica, con un pronóstico excelente. Presentamos dos pacientes con GnS diagnosticados e intervenidos en nuestro centro hospitalario.
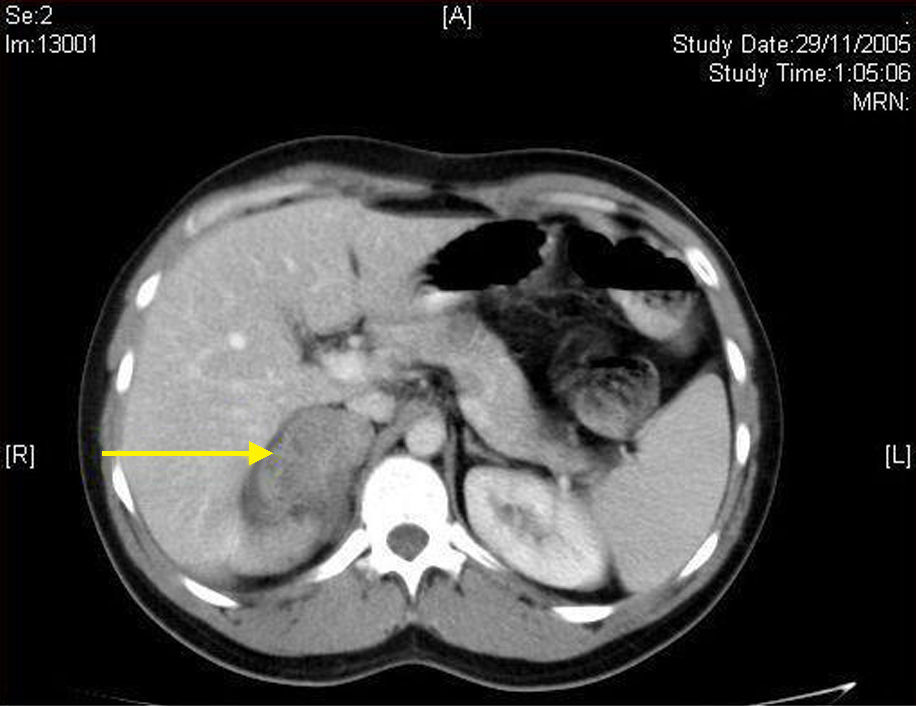
Caso 1. Se trata de una mujer de 18 años de edad sin antecedentes de interés, que siendo estudiada de forma ambulatoria por un dolor abdominal inespecífico se le encontró en un estudio ecográfíco una masa sólida suprarrenal izquierda. La tomografía computarizada (TC) abdominal confirmó la presencia de una tumoración suprarrenal de 6 cm de diámetro, homogénea, sólida y con realce periférico. Los estudios de laboratorio y despistaje funcional hormonal fueron normales. Con el diagnóstico de masa suprarrenal no funcionante se indicó cirugía, que consistió en una suprarrenalectomía izquierda por abordaje laparoscópico. La paciente fue dada de alta a las 24 h de la intervención y la pieza quirúrgica informó de tumoración de 7 cm con diagnóstico anatomopatológico de Gn.
Caso 2. Es el caso de un varón de 28 años de edad sin antecedentes de interés salvo episodios recurrentes de gastroenteritis y cuadros inespecíficos de dolor abdominal. En una ecografía realizada de forma ambulatoria se informó de la sospecha de una tumoración suprarrenal derecha, confirmando la TC abdominal la presencia de una masa suprarrenal de 5,8 × 4 cm de densidad mixta baja. Los análisis rutinarios y el perfil hormonal en sangre fueron, como en el caso anterior, normales. Con el diagnóstico preoperatorio de tumor incidental suprarrenal no funcionante se realizó una suprarrenalectomía derecha laparoscópica. El paciente fue igualmente dado de alta a las 24 h sin complicaciones postoperatorias y la anatomía patológica indicó que se trataba de un ganglioneuroma.
El Gn es un tumor benigno, raro y de crecimiento lento originado a partir de las neuronas simpáticas primitivas de la cresta neural. Pertenece al grupo de los tumores neurogénicos originados a este nivel, junto al ganglioblastoma y el neuroblastoma, y a diferencia de ellos está formado por células ganglionares maduras sin potencial maligno1–5. Existe controversia sobre si la aparición de un Gn puede ocurrir de novo (primario) o si es resultado de la diferenciación y maduración a partir de ganglioblastomas o neuroblastomas3,4. Aparecen a lo largo de la cadena simpática paravertebral que va desde el cuello a la pelvis y ocasionalmente en la medula adrenal, siendo sus localizaciones más frecuentes mediastino posterior (40%) y retroperitoneo (37%); más rara es su presentación en mediastino anterior, estómago, apéndice o próstata1–5. La localización suprarrenal se estima en un 15-30% de los casos.
La incidencia de GnS ha crecido en los últimos años en relación directa con el incremento en la detección de incidentalomas por la difusión de estudios de imagen que cada vez son de mayor calidad. Se estima que entre el 1 y el 10% de las TC abdominales encuentra de forma incidental un tumor adrenal, de los que el 1-6% corresponde a Gn1. Aunque también se han notificado casos en niños, el GnS afecta fundamentalmente a adultos jóvenes sin predominio de sexo1,2. Aproximadamente la mitad de los pacientes está asintomática, y cuando se manifiestan, la clínica más común es de dolor abdominal inespecífico o masa palpable. Desde el punto de vista funcional son, en general, no secretores, aunque en un 20-30% de los casos producen catecolaminas y metabolitos. En caso de actividad hormonal puede acompañar diarrea (liberación de péptido intestinal vasoactivo), sudoración o hipertensión arterial, pero sin producir emergencias clínicas, a diferencia del feocromocitoma4.
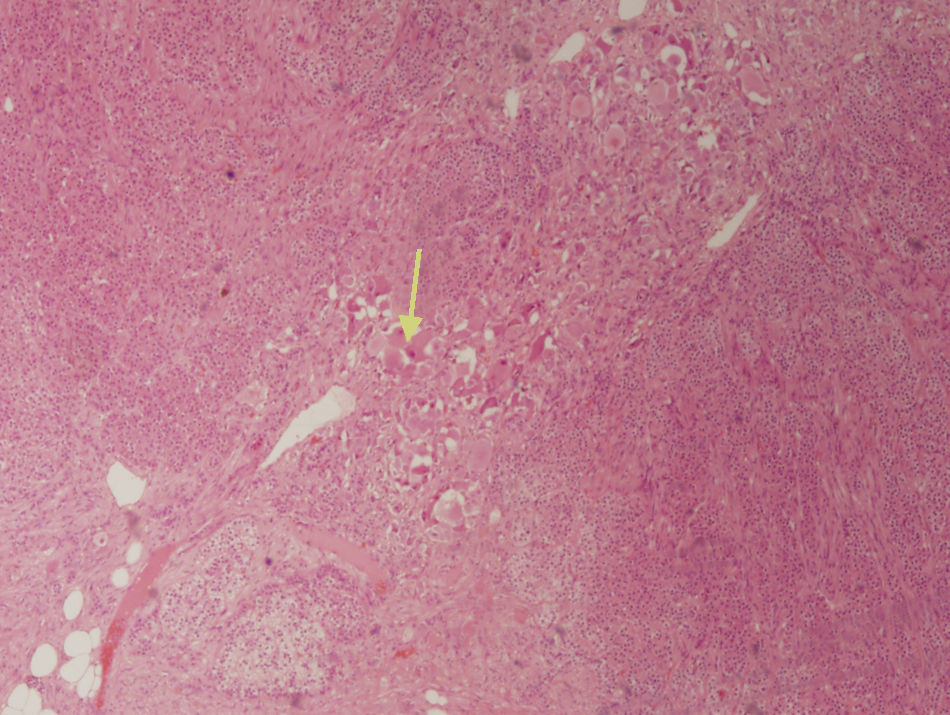
El diagnóstico comienza siempre con un estudio hormonal que demuestre no funcionalidad del tumor: cortisol libre en orina de 24 h, supresión con 1 mg de dexametasona, cortisol basal en sangre, ACTH, renina, aldosterona en sangre y catecolaminas (adrenalina y noradrenalina) y sus metabolitos en sangre y orina3. Las pruebas de imagen más útiles son la TC abdominal y la resonancia magnética nuclear (RMN), que han demostrado su superioridad respecto de la ecografía en la detección y caracterización de la patología suprarrenal1. En la TC se presentan como una masa sólida hipoatenuada (habitualmente, menos de 40 unidades Hounsfield) y bien definida, de forma oval o lobulada y con cápsula fibrosa (fig. 1), presentando del 42 al 60% calcificaciones intratumorales; en ocasiones, pueden ser heterogéneas tras la administración de contraste2,3,5,6. En la RMN presentan una señal de intensidad baja en T1 y heterogeneidad con intensidad alta en T25. Recientemente se ha incorporado la PET para complementar ambas pruebas en el diagnóstico del Gn y con la intención, sobre todo, de descartar patología neoplásica maligna suprarrenal1. El diagnóstico de certeza definitivo vendrá determinado tras el análisis histopatológico de la pieza quirúrgica. En el estudio microscópico es característico observar una imagen uniforme con un estroma constituido por células de Schwann orientadas transversal y longitudinalmente, que se entrecruzan de forma irregular, pudiéndose encontrar en ocasiones grasa. Dispersas por este fondo schwanniano se encuentran neuronas relativamente maduras, con escasa sustancia de Nissl y formando pequeños grupos o nidos. Es típico el citoplasma voluminoso eosinófilo y la presencia de uno a tres núcleos con atipia ligera o moderada (fig. 2). El uso de la punción aspiración con aguja fina está limitado por la posibilidad de que se trate de patología maligna (carcinoma adrenal o metástasis) o una lesión quística y la dificultad para diferenciar entre adenoma y carcinoma2,3.

 con un estroma neuromatoso, creciendo sobre tejido suprarrenal normal.")
La indicación quirúrgica en tumores adrenales incidentales no funcionantes no está del todo definida y va a depender del tamaño de la lesión y sus características radiológicas. No hay duda en aquellos tumores que asocian síntomas, tienen un diámetro mayor de 6 cm (se ha visto una mayor incidencia de carcinoma en estas lesiones) o presentan características radiológicas de malignidad3,7. En lesiones menores de 4 cm suele hacerse vigilancia clínico-radiológica, aunque algunos autores abogan por exéresis en pacientes jóvenes en vistas a un seguimiento muy largo y a la ansiedad que puede generarles7,8. La controversia se establece para lesiones entre 4 y 6 cm, aceptándose tanto realizar exéresis como seguimiento, planteándose entonces la cirugía en caso de crecimiento del tumor o aparición de signos radiológicos de malignidad3,7.
La laparoscopia se ha convertido en la vía de abordaje de elección para toda la patología suprarrenal, incluyendo las masas incidentales no funcionantes y con cada vez menor limitación por el tamaño8. No existe consenso actual sobre el tamaño máximo del tumor que se debe intervenir por laparoscopia, estando el límite clásico aceptado en 6 cm; existen, no obstante, numerosas series con tumores de mayor tamaño, llegando incluso hasta los 13 cm1,7–10. La vía de abordaje suele ser transperitoneal, aunque hay grupos que prefieren el acceso retroperitoneal, siendo contraindicaciones absolutas, hoy día, la presencia de tumor invasivo maligno o la trombosis venosa asociada de la vena renal o suprarrenal7–10. El pronóstico del GnS tras la resección es excelente, incluso cuando una cirugía exerética completa no es posible11; la recurrencia es excepcional, debiendo interpretarse entonces como una cirugía inicial incompleta (sería entonces, más bien, «persistencia»).
