


Los gangliocitomas hipofisarios son tumores benignos poco frecuentes (menos de un centenar de casos descritos hasta el momento)1–6, con bajo potencial de proliferación y crecimiento lento.
Histológicamente la mayoría son tumores mixtos compuestos por célula ganglionares neuronales y una proliferación adenomatosa de células adenohipofisarias4,7,8; los 2 componentes pueden aparecer como un único nódulo o coexistir como tumores separados pero adyacentes. La mayor parte de los casos se asocian a hiperproducción hormonal, siendo la GH la hormona más frecuentemente producida3 y en casos más raros prolactina1,2 o ACTH, aunque también existen tumores no funcionantes como en el caso que se describe a continuación.
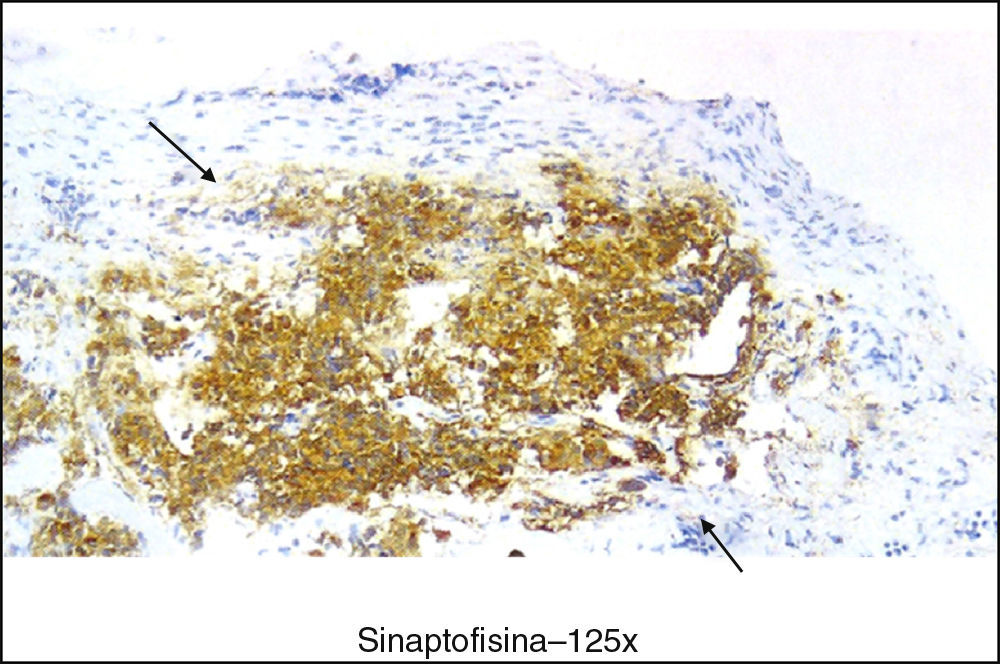
Se trata de un varón de 43 años, afecto de hipertensión arterial y con tuberculosis ganglionar intervenida a los 28 años. En sus antecedentes familiares destaca que su padre ya fallecido (por aneurisma de aorta) tuvo un adenoma hipofisario no productor, con expansión supraselar diagnosticado a los 60 años. El paciente había consultado por cefalea occipital intensa, comprobándose en la resonancia cerebral la existencia de una lesión redondeada en la vertiente lateral derecha de la hipófisis, de 10mm de diámetro máximo con elevación del diafragma selar, hiperintensa en T2 que no se realzaba con el contraste, compatible con adenoma hipofisario. No presentaba clínica de disfunción hipofisaria ni de hipopituitarismo. En la exploración física destacaba una obesidad grado 2 (índice de masa corporal 35kg/m2), siendo el resto anodino. Se realizó determinación de hormonas adenohipofisarias y de órganos diana basales, siendo todas normales, salvo cortisol basal (8:30 horas) de 30μg/dl y ACTH basal de 72 pg/ml (rango normal: 0-46 pg/ml); las cortisolurias de 24 horas fueron normales y el cortisol plasmático tras 1mg de dexametasona fue inferior a 2μg/dl. El paciente fue intervenido por vía transesfenoidal realizándose una exéresis completa; la anatomía patológica describió un tumor neuroepitelial constituido por células ganglionares maduras distribuidas en grupos de neuronas bipolares, con estroma de células gliales no neoplásicas y fibras de reticulina. Las células ganglionares eran positivas inmunohistoquímicamente para sinaptofisina (fig. 1) y cromogranina; las células gliales se teñían con proteína glial fibrilar. No se objetivó proliferación adenomatosa. Tras la cirugía el paciente permaneció asintomático y sin datos radiológicos de restos tumorales ni recidiva; tampoco presentaba clínica de hipercortisolismo.

Los gangliocitomas son lesiones infrecuentes en la silla turca que habitualmente se asocian a hipersecreción de hormonas hipofisarias1-4 (fundamentalmente GH y en menor medida prolactina y ACTH) aunque también pueden ser endocrinológicamente no funcionantes como en este caso. Su coexistencia con adenomas hipofisarios se da en el 0,52% de las lesiones de la silla turca5. La mayoría de los casos descritos ocurren en mujeres6.
El origen de estos tumores es un motivo de controversia y existen distintas hipótesis. La primera consiste en que el adenoma hipofisario es el resultado de una estimulación paracrina o endocrina por parte de la hormona hipotalámica liberadora de hormona hipofisaria, producida por las células ganglionares heterotópicas e intraselares sobre las células adenohipofisarias (aunque esto no explicaría por qué se forma un adenoma y no una hiperplasia)5. La segunda teoría sostiene que los gangliocitomas se forman debido a la diferenciación neuronal de células de un adenoma escasamente granulado, aunque esta opción no explica todos los tipos de tumores2. La tercera apunta a que el origen son restos embrionarios que contienen células con características intermedias entre células ganglionares y células adenohipofisarias8; es la hipótesis más probable ya que es la que mejor explica las características tanto de los tumores mixtos como de los ganglionares puros.
En base a los hallazgos clínicos, bioquímicos y radiológicos estas lesiones son indistinguibles de un adenoma, por lo que el diagnóstico se obtiene tras la cirugía con el estudio histológico. El estudio inmunohistoquímico con marcadores neuronales y gliales (sinaptofisina y enolasa neuronal específica principalmente) y anticuerpos contra las hormonas hipofisarias, confirma el diagnóstico. Es útil también el uso de estudios ultraestructurales.
La actitud terapéutica a seguir debe ser la misma que ante un adenoma.
