



Con la mayor utilización actual de las pruebas diagnósticas radiológicas hay una incidencia del 4,3% (1,5-7%)1 de incidentaloma suprarrenal. Se ha estimado que hasta un 10% de éstos son feocromocitomas.
A continuación presentamos un caso de incidentaloma suprarrenal en una paciente afecta de síndrome pluriglandular autoinmunitario tipo II (SPGA): enfermedad de Addison autoinmunitaria, hipotiroidismo primario autoinmunitario y vitíligo, lo que constituye una rareza clínica.
CASO CLÍNICOMujer de 78 años, con antecedentes personales de sordera neurosensorial congénita y de SPGA tipo II. Fue diagnosticada de enfermedad de Addison a los 46 años, de vitíligo (que afecta a manos, aréolas, codos y rodillas) a los 48 años y de hipotiroidismo primario autoinmunitario a los 66 años.
Además presenta diabetes mellitus tipo 2 desde los 67 años, en tratamiento con antidiabéticos orales (sulfonilureas) hasta hace un año, que ha precisado tratamiento insulínico. Como complicaciones presenta retinopatía diabética preproliferativa y afectación neuropática vegetativa con gastroparesia.
Asimismo, presentó un episodio de infarto agudo de miocardio a los 72 años.
Se encuentra en tratamiento con levotiroxina (50 mg), hidrocortisona (30 mg), insulina premezclada 30/70, ácido acetilsalicílico y omeprazol.
La paciente ingresa en el servicio de endocrinología, por presentar mal control glucémico (cifra de glucohemoglobina del 10,4%), además refería intensa astenia, sensación nauseosa y malestar en el desayuno, que no cede tras la in-gesta, así como síntomas vegetativos con el paso de decúbito a bipedestación. Se realiza análisis de sangre donde se objetiva anemia ferrópenica.
Exploración física: presión arterial (PA), 130/70 mmHg en decúbito y 100/50 mmHg en ortostatismo; frecuencia cardíaca, 90 lat/min; temperatura, 36 °C; consciente y orientada, eupneica, con palidez cutaneomucosa. No se palpa bocio, no se escucha soplo carotídeo. Auscultación cardiopulmonar dentro de límites normales. Abdomen blando, depresible, no se palpan masas ni megalias, peristaltismo conservado, no doloroso a la palpación.
Extremidades inferiores: pulsos pedios presentes, sin signos de trombosis venosa profunda.
Durante el ingreso se realizan diversas exploraciones complementarias para estudio y valoración del cuadro clínico.
En la gastroscopia se visualiza un cardias incompetente y un pólipo gástrico, en la colonoscopia un pólipo rectal y motilidad de colon espástica.
En el estudio ginecológico no se detectó ningún cuadro patológico.
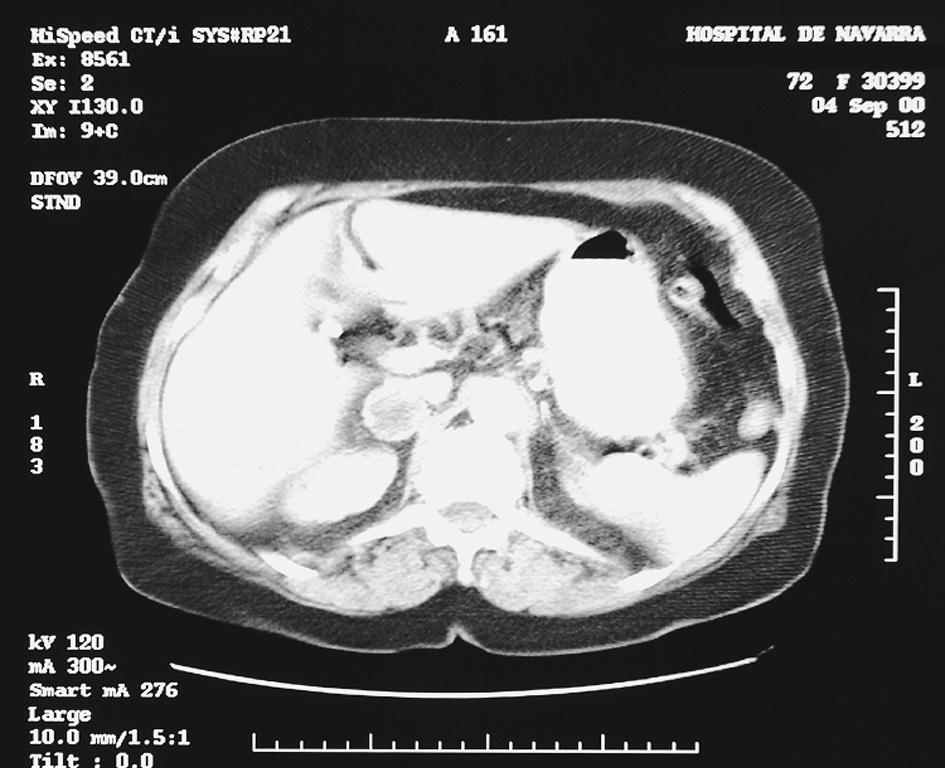
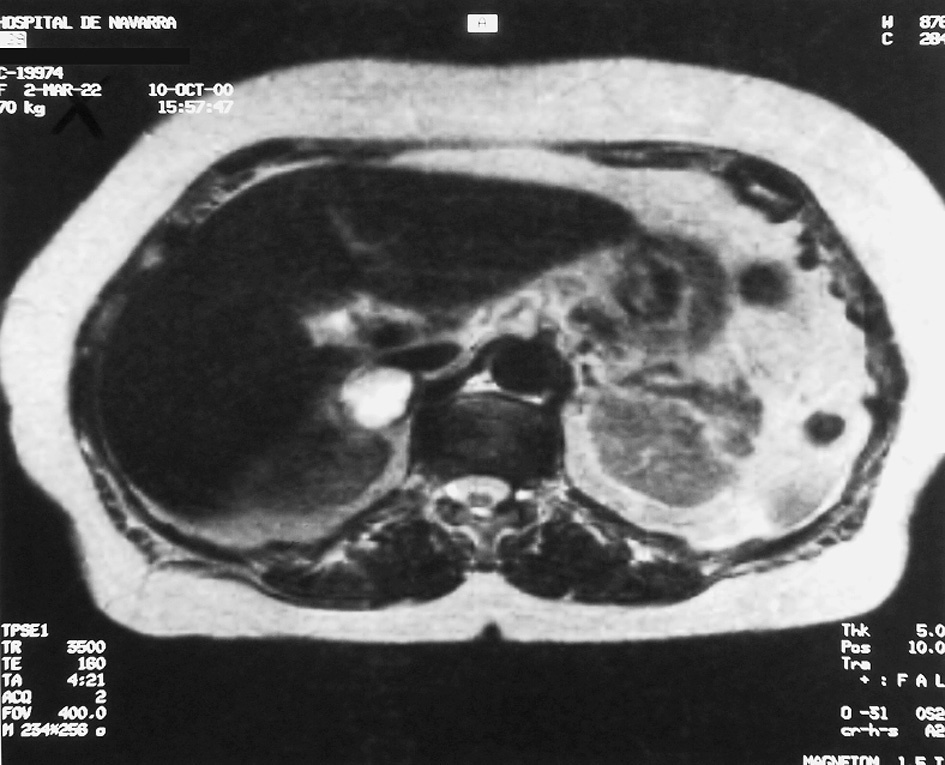
Se realiza tomografía computarizada (TC) abdominal (fig. 1), en la cual se objetiva una masa de aspecto heterogéneo con aparente zona de necrosis en la glándula suprarrenal derecha, de 3 cm de diámetro. Se completa el estudio con resonancia magnética (RM) abdominal (fig. 2), donde se delimita una masa de 26 mm de diámetro mayor horizontal y 36 mm de diámetro mayor vertical, de intensidad heterogénea, con áreas hiperintensas en T2 e intensidad de señal similar en imágenes en gradiente en fase y fuera de fase.
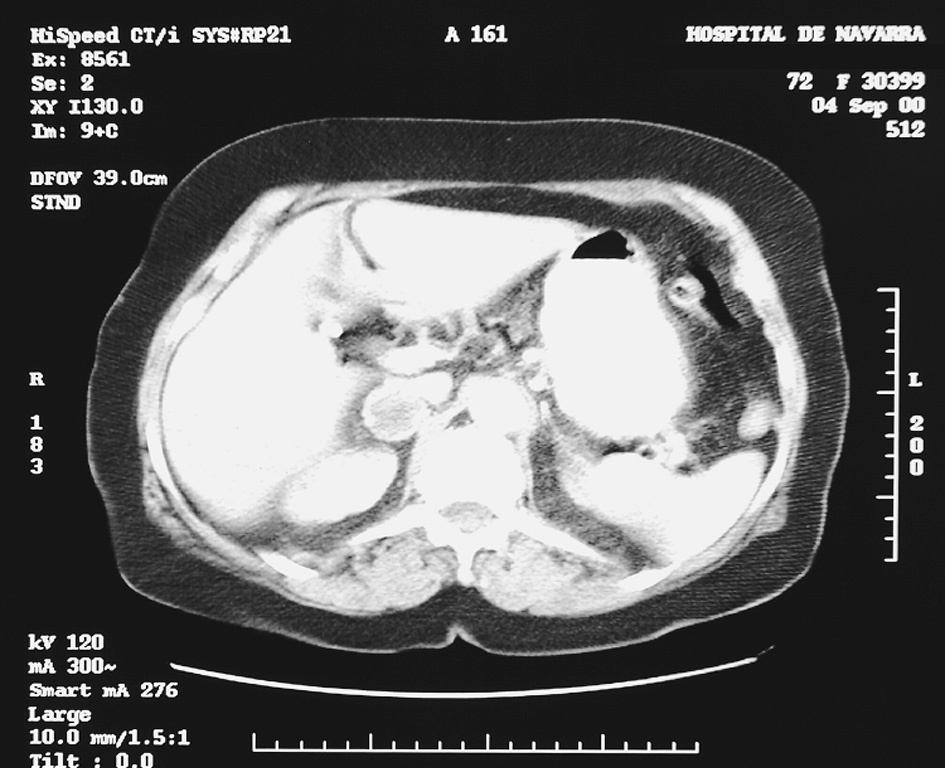
Fig. 1. Tomografía computarizada que muestra una masa de aspecto heterogéneo con aparente zona de necrosis, de 3 cm de diámetro, en la glándula suprarrenal derecha.
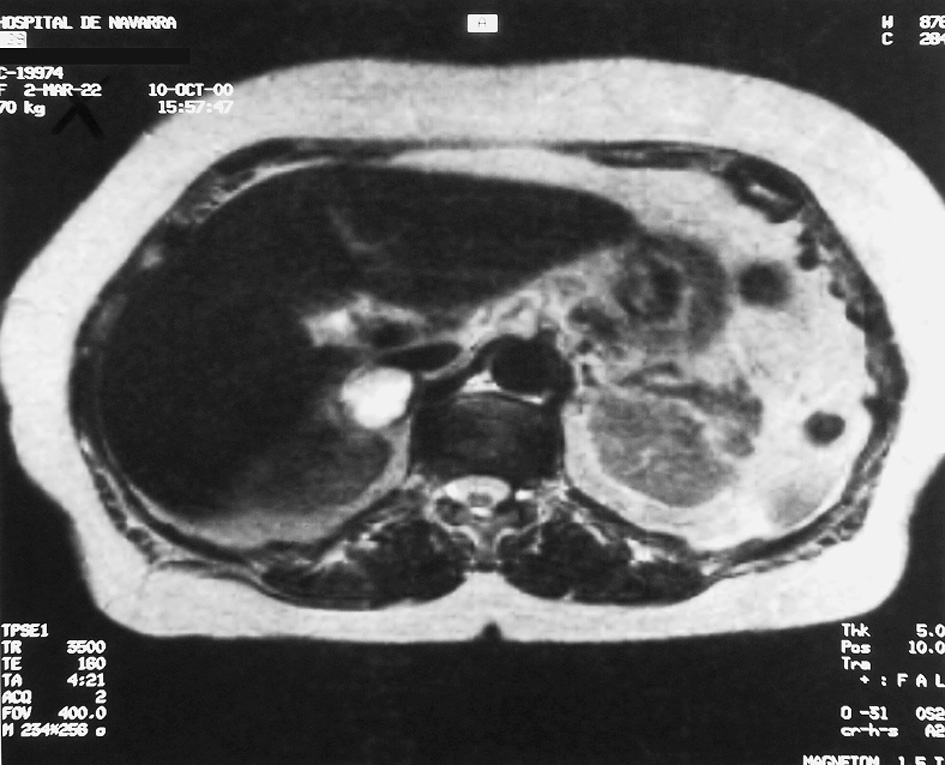
Fig. 2. Resonancia magnética en la que se observa una masa suprarrenal derecha de 26 µ36 mm, de intensidad de señal moderadamente hererogénea, similar a la del parénquima hepático de T1, e hiperintensa en T2.
Ante la sospecha radiológica de feocromocitoma, se realiza análisis de orina para cuantificar metanefrinas y catecolaminas fraccionadas, las cuales presentan concentraciones hasta 10 veces mayores que los valores de referencia (tabla 1).

TABLA 1. Valores de metanefrinas y catecolaminas en orina de 24 horas
Se realiza una gammagrafía con metayodobencilguanidina (MIBG), con yodo 131, que no presenta captación.
Ante el diagnóstico de sospecha, clínico, analítico y por prueba de imagen (TC y RM) de feocromocitoma, se comienza con la preparación prequirúrgica, por lo que se instaura tratamiento con fenoxibenzamina, con mala tolerancia al principio, con episodios de hipotensión, que mejoran progresivamente, y se puede llegar a dosis máximas de 20 mg/día. No precisa tratamiento con bloqueadores beta, ya que presenta buen control de la frecuencia cardíaca.
Dada la edad de la paciente y al no presentar antecedentes familiares de síndromes asociados al feocromocitoma, no se solicita estudio genético2.
Se intenta intervención quirúrgica por vía laparoscópica, pero debido a múltiples adherencias secundarias a colecistectomía previa, se procede a realizar la intervención mediante laparotomía subcostal derecha sin incidencias.
Presenta un postoperatorio sin complicaciones. Se repite la determinación de catecolaminas y metanefrinas urinarias tras la intervención con normalización de sus valores.
La paciente presenta mejoría clínica significativa, con desaparición de los síntomas nauseosos y de la hipotensión ortostática.
DISCUSIÓNEl feocromocitoma es un tumor del sistema nervioso simpático que se desarrolla a partir de las células cromafines, y que se caracteriza por la producción excesiva de catecolaminas. El 85% de los casos se localizan en la médula adrenal y el 15% restante son extraadrenales (paragangliomas). Presenta una incidencia de 0,8/100.000 habitantes/año.
La clínica del feocromocitoma puede ser muy variada: los síntomas más comunes son cefalea, diaforesis y palpitaciones. Causa hasta el 0,2% de los casos de hipertensión arterial (HTA). Otros signos que se pueden presentar son palidez cutánea, hipotensión ortostática, visión borrosa, hiperglucemia, leucocitosis y aumento de velocidad de sedimentación globular.
Hay distintas formas de presentación: con síntomas clásicos, iniciarse como miocardiopatía catecolaminérgica (edema agudo de pulmón), como síntomas gastrointestinales o incluso como trastornos psiquiátricos. Hasta el 40% son asintomáticos.
Hay distintos factores desencadenantes de la clínica, como el ejercicio físico, la inducción anestésica o las exploraciones endoscópicas3-5.
Para el diagnóstico la cuantificación de metanefrinas y catecolaminas fraccionadas en orina es una prueba muy específica y sensible (del 98% y el 90%). La medición de las metanefrinas en plasma tiene mayor sensibilidad (99%), pero menor especificidad (82%), por lo que se suele reservar para casos con un alto índice de sospecha6-8.
En lo referente a la confirmación radiológica, la TC presenta una sensibilidad del 98,9%, y con la RM se llega hasta el 100%9.
La gammagrafía con MIBG tiene una sensibilidad del 81 al 95%. Si se realiza con yodo 131 hay una tasa de falsos negativos del 13-25%, este porcentaje disminuye con el uso de yodo 123, el cual es menos accesible por su mayor coste económico4.
El tratamiento es quirúrgico10, siempre que sea posible. Es conveniente utilizar bloqueadores alfa unos 7-10 días antes de la intervención quirúrgica, hasta alcanzar cifras de PA en decúbito menores de 160/90 mmHg, y en ortostatismo, mayores de 80/45 mmHg. Se debe realizar electrocardiograma, en el cual no debe aparecer más de un extrasístole ventricular cada 5 s, además no debe haber alteraciones en el segmento ST11. Para conseguir el control de la frecuencia cardíaca, se pueden utilizar bloqueadores beta, siempre que se haya conseguido una inhibición alfa adecuada.
En nuestra paciente afecta de SPGA tipo II12, con insuficiencia adrenal primaria autoinmunitaria, ha aparecido un tumor de las células de la médula adrenal. No hay descrito ningún tipo de relación causal entre ambos procesos, por lo que resulta cuando menos un hallazgo anecdótico.
Se midieron anticuerpos anticápsula suprarrenal que fueron negativos, lo cual puede explicarse porque en el momento de la medición habían transcurrido 22 años desde el diagnóstico de la enfermedad. Los anticuerpos antitiroideos fueron positivos y los anticuerpos contra la célula beta del páncreas (antiácido glutámico descarboxilasa y antiinsulina) fueron negativos, ambos medidos en el momento del diagnóstico de las respectivas afecciones.
Existen diversas causas de insuficiencia adrenal primaria (enfermedad de Addison)13-15, en todas ellas, exceptuando la adrenalitis autoinmunitaria, se produce una destrucción total de la glándula suprarrenal, que afecta tanto a la corteza como a la médula. Sin embargo, en la adrenalitis autoinmunitaria la médula adrenal está preservada, lo cual ha permitido, en este caso, el desarrollo de un tumor dependiente de ésta.
Esta curiosa asociación de enfermedad suprarrenal autoinmunitaria en el contexto de SPGA tipo II y tumor de la médula adrenal (feocromocitoma) no se había descrito hasta el momento.
Correspondencia: Dra. M. Toni. Servicio de Endocrinología. Hospital de Navarra. Irunlarrea, 3. 31008 Pamplona. Navarra. España. Correo electrónico: mtonigar@hotmail.com
Manuscrito recibido el 24-6-2008 y aceptado para su publicación el 8-9-2008.
