


Los síndromes de osteólisis hereditaria se caracterizan por destrucción esquelética debida a una incontrolada resorción ósea. Constituyen un grupo heterogéneo de entidades infrecuentes de clasificación aún discutida, en la que se incluyen formas multicéntricas carpotarsales: síndromes de Winchester, de nodulosis-artropatía-osteólisis (NAO) y de Torg, de herencia autosómica recesiva. Se presenta el caso de un niño de 13 años y origen marroquí, con incapacidad para la bipedestación y la manipulación por deformidad progresiva de las manos y los pies, así como fracturas patológicas ante mínimos traumatismos. Radiológicamente faltan carpo y tarso, y hay fracturas en las extremidades y osteoporosis intensa, sin alteraciones en el metabolismo fosfocálcico. Presenta alteraciones corneales y agudeza visual muy disminuida. Su único hermano tiene los mismos síntomas; sus padres son primos y están sanos. Ante tales hallazgos, se establece el síndrome de Winchester como diagnóstico más probable.
Hereditary osteolysis syndromes are characterized by the destruction of skeletal groups produced by uncontrolled bone resorption. These syndromes constitute a heterogeneous group of infrequent entities. The classification of these entities is still controversial and includesthe following multicentric carpal-tarsal forms: Winchester, NAO (nodulosis-arthropathy-osteolysis) and Torg syndromes, all of which show autosomal recessive inheritance. We present a 13-year-old boy of Moroccan origin with impaired walking and hand function due to progressive deformity of the hands and feet, as well as pathological fractures after minimal trauma. Radiographic examination revealed dissolution of carpal and tarsal bones, fractures in the extremities and intense osteoporosis, without alterations in phosphocalcic metabolism. The patient had corneous alterations and highly diminished visual keenness. The boy’s only brother showed the same symptoms but his parents and cousins were healthy. Based on the findings in our patient, Winchester syndrome was established as the most probable diagnosis.
Los síndromes de osteólisis hereditaria son un grupo heterogéneo de entidades caracterizadas por la destrucción de grupos óseos, resultado de una resorción ósea incontrolada. Su incidencia es muy baja y son escasos los casos descritos en el mundo1,2. A pesar de que su clasificación es aún controvertida, prevalece la propuesta por la Sociedad Internacional de Displasias Esqueléticas (ISDS)1, según la cual las osteólisis carpotarsales de herencia autosómica recesiva son los síndromes de Winchester (SW), de nódulos, artropatía y osteólisis (NAO) y de Torg (ST), entidades no siempre fáciles de diferenciar.
Presentamos el caso de un niño de 13 años cuyas deformidades progresivas asociadas a la ausencia bilateral de carpo y tarso, contracturas articulares y osteoporosis generalizada nos hicieron sospechar el diagnóstico de SW.
CASO CLÍNICOVarón de 13 años y origen marroquí que es remitido a nuestro hospital para estudio y tratamiento de deformidades óseas sin filiar. Desde la primera infancia sufre dolor articular en las cuatro extremidades, de predominio distal y sin inflamación asociada. Presenta deformidad progresiva de manos y pies que impide su bipedestación en los últimos 2 años y dificulta intensamente la manipulación, lo que afecta al desarrollo de actividades sencillas de la vida diaria. Asimismo refiere fracturas por traumatismos de baja intensidad: en tibia izquierda 1 año antes y dos fracturas en fémur derecho 11 meses y 8 meses antes, respectivamente. Las tres precisaron inmovilización con yeso, y no ha recibido otro tratamiento para su enfermedad hasta su llegada a nuestro hospital.
No se conoce otros antecedentes personales, ya que procede de un centro de acogida. Refiere que sus padres son primos, de origen marroquí, sin clínica alguna. Sin embargo, su único hermano, de 20 años, tiene igual sintomatología.
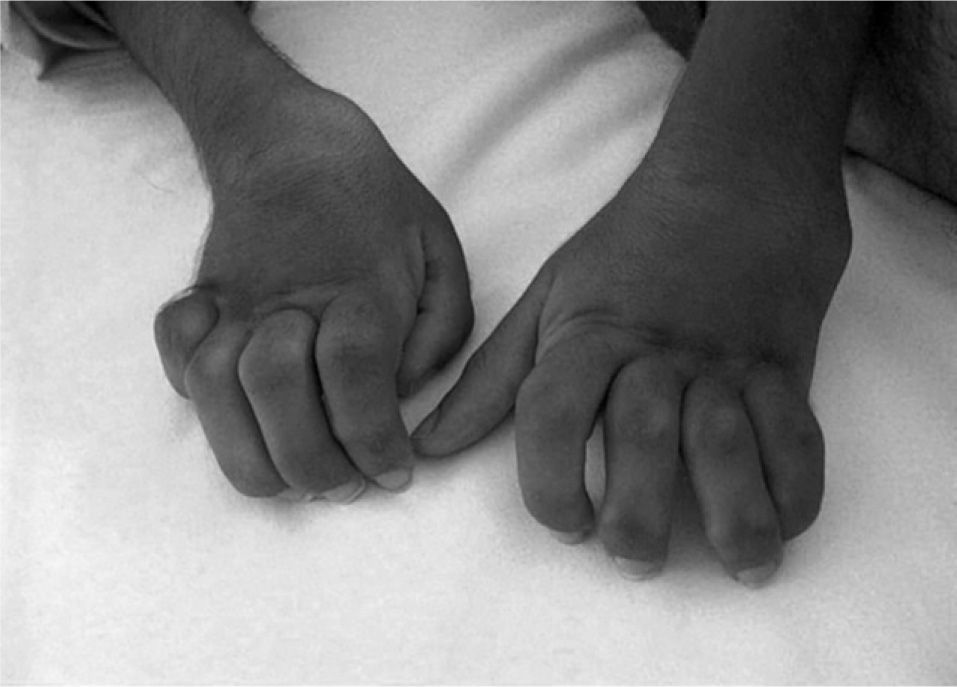
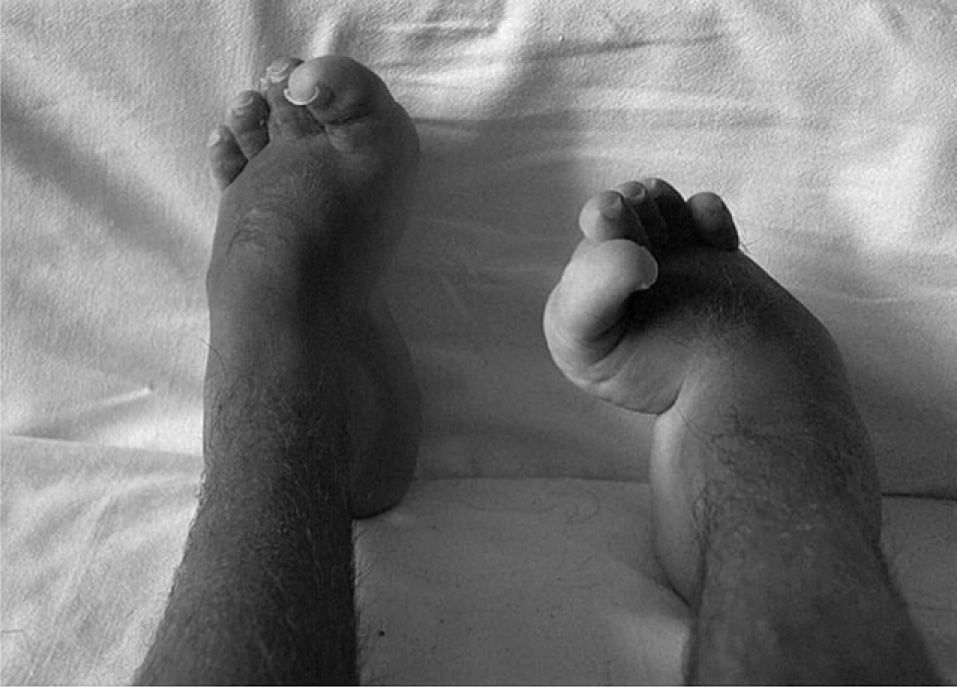
Exploración físicaSegún las gráficas de Tanner, presentaba peso (25kg) bajo el percentil 3 y talla (145cm) en percentil 3–10 para su sexo y edad. Buen estado general, normocoloreado. Facies tosca, sin rasgos dismórficos. Hipertricosis en las cuatro extremidades sin otras alteraciones cutáneas. La auscultación cardiopulmonar y la exploración abdominal fueron normales. El desarrollo puberal correspondía a estadios de Tanner II-III. La exploración neurológica fue normal, aunque sin marcha autónoma, por apoyo plantar inestable. En el aparato locomotor destacaban una intensa atrofia muscular generalizada, cifosis torácica, deformidad e incapacidad para extensión completa de ambos codos, flexo de cadera y rodilla derechas, manos en garra (fig. 1) y pies cavos en adducto (fig. 2). No presentaba signos inflamatorios en las articulaciones.
Exploraciones complementarias

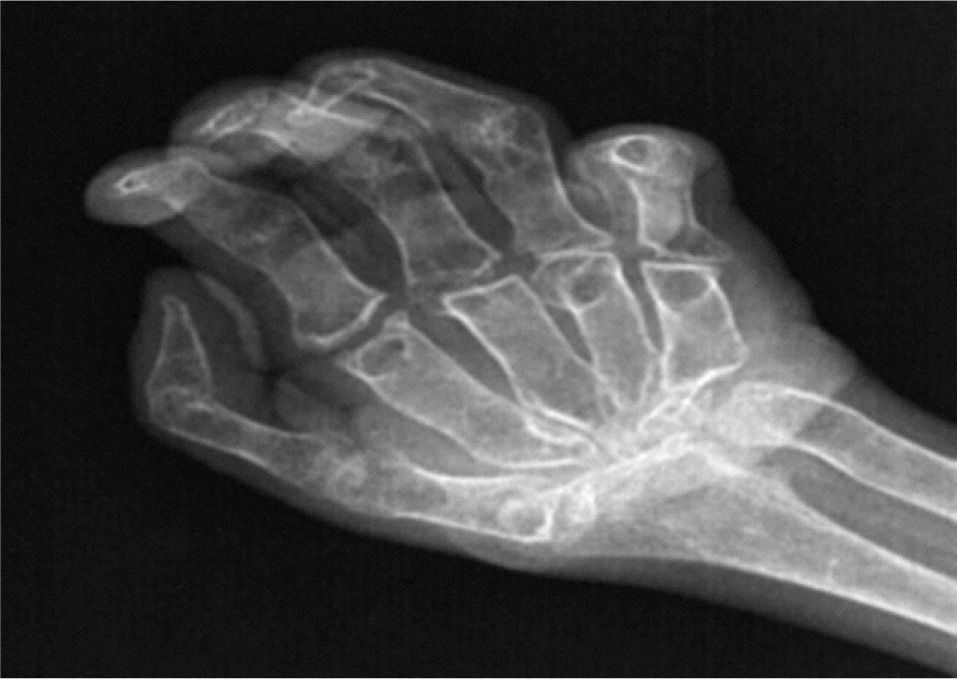
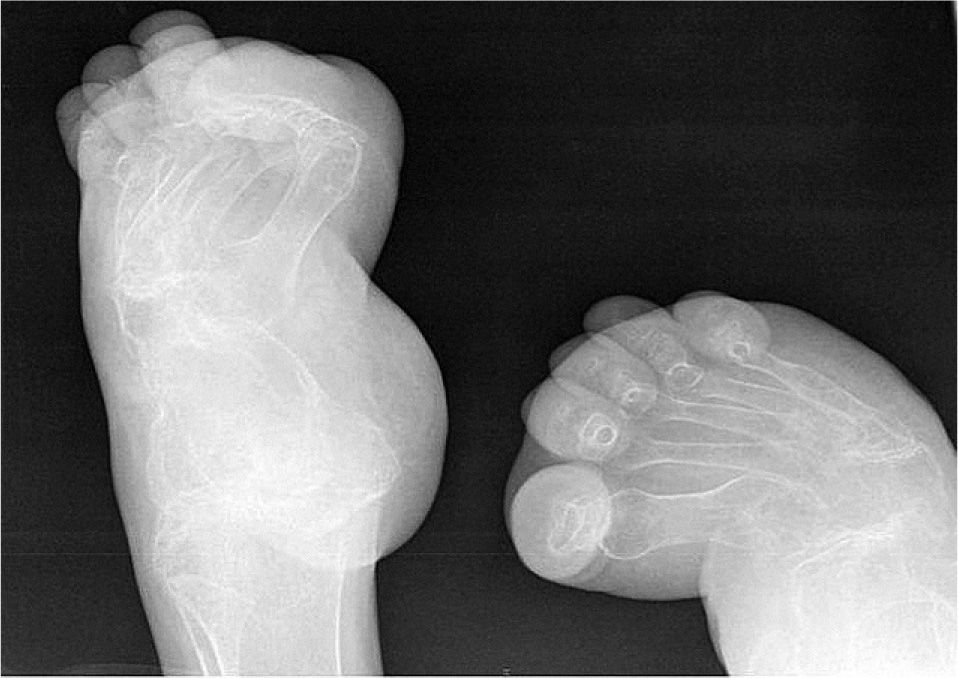
El hemograma y la bioquímica en sangre y orina estaban dentro de la normalidad. Velocidad de sedimentación globular en la primera hora, 22mm/h. Metabolismo fosfocálcico en sangre y orina, normal. Paratirina (PTH) intacta, 24pg/ml (10–65pg/ml), y 25(OH)-vitamina D, 45ng/ml (16- 74ng/ml), dentro del rango de referencia. Electromiografía: sin alteraciones. La serie ósea mostró una intensa osteopenia generalizada, ausencia de carpos y sinóstosis interfalángica en las manos (fig. 3), incurvación de fémur derecho con callos de fracturas previas, fractura-luxación de codos, vértebras dorsales bicóncavas y tarsos incompletos, con cuñas no identificables (fig. 4). En la densitometría ósea lumbar (L2-L4), osteoporosis intensa (Z-score=−4,3). En la exploración oftalmológica la agudeza visual bilateral fue del 10%; se observó queratocono bilateral, y disminución de la presión intraocular y del diámetro de ambos ojos.


Ante la clínica y los resultados del estudio, se estableció el síndrome de Winchester como posible diagnóstico de sospecha.
Los objetivos iniciales del tratamiento fueron aumentar la masa ósea y evitar el dolor del paciente, ya que sólo se podrán corregir quirúrgicamente las deformidades esqueléticas si se corrige la osteoporosis. Por ello se administraron ciclos de pamidronato intravenoso (1mg/kg/día, durante 3 días, cada 4 meses) y analgesia oral. No le fue concedido el tratamiento con hormona de crecimiento recombinante, solicitado para aumentar la masa muscular, facilitar la ganancia ósea y aliviar el dolor. Se realizó control densitométrico cada dos ciclos de terapia y se recomendó fisioterapia en su ciudad de origen.
Desde el punto de vista oftalmológico, se trató el queratocono con lentillas y se realizó seguimiento periódico, ya que no se descarta la necesidad de cirugía.
DISCUSIÓNLos cuadros de osteólisis son un grupo heterogéneo de entidades con diferentes manifestaciones clínicas y con base fisiopatológica común que consiste en una incontrolada resorción ósea que destruye grupos esqueléticos diversos. Su clasificación es aún objeto de debate, aunque la más aceptada es la propuesta por la Sociedad Internacional de Displasias Esqueléticas (ISDS) en 20011.
De las osteólisis, la multicéntrica de predominio carpotarsal e interfalángico es la que nos ocupa e incluye los síndromes de NAO, Torg o Winchester, ya que los tres son de herencia autosómica recesiva, compatibles con nuestro caso. Todos ellos son muy infrecuentes y hasta el momento hay muy pocos casos descritos en el mundo y no hay ninguno publicado en España1,2.
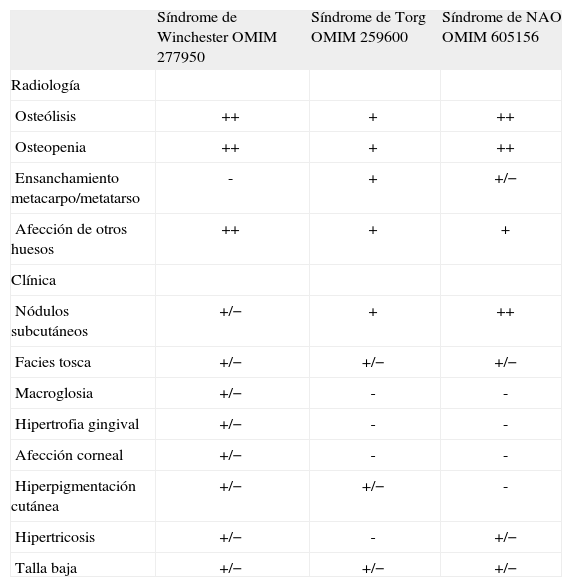
En los tres cuadros la osteólisis es idiopática, comienza a nivel interfalángico y carpotarsal y puede generalizarse después. Se diferencian entre sí por la gravedad de la osteólisis y por los hallazgos asociados a ella2,3 (tabla 1). Algunos autores señalan que podría tratarse de procesos con igual base genética y diferente expresión fenotípica3,4, ya que tanto en casos de SW como de ST se ha descubierto una misma mutación en homocigosis en el gen que codifica una enzima implicada en la resorción ósea, la metaloproteinasa 2 de la matriz, y se sitúa en el cromosoma 16 (16q12-21)4,5. El estudio genético no ha podido realizarse en la actualidad por no disponerse de esta prestación en los laboratorios nacionales y otros.
Características clinicorradiológicas de las formas osteolíticas multicéntricas autosómicas recesivas
| Síndrome de Winchester OMIM 277950 | Síndrome de Torg OMIM 259600 | Síndrome de NAO OMIM 605156 | |
| Radiología | |||
| Osteólisis | ++ | + | ++ |
| Osteopenia | ++ | + | ++ |
| Ensanchamiento metacarpo/metatarso | - | + | +/− |
| Afección de otros huesos | ++ | + | + |
| Clínica | |||
| Nódulos subcutáneos | +/− | + | ++ |
| Facies tosca | +/− | +/− | +/− |
| Macroglosia | +/− | - | - |
| Hipertrofia gingival | +/− | - | - |
| Afección corneal | +/− | - | - |
| Hiperpigmentación cutánea | +/− | +/− | - |
| Hipertricosis | +/− | - | +/− |
| Talla baja | +/− | +/− | +/− |
-: ausente; +/−: ocasionalmente presente; +: presente; ++: prominente; NAO: síndrome de nodulosis, artropatía y osteólisis.
El temprano inicio, la evolución progresiva y posterior estabilización durante la adolescencia de nuestro caso hacen sospechar los tres cuadros referidos. Por otra parte, las artralgias son permanentes y comunes a los tres6,7. La resorción ósea conlleva osteopenia, generalmente intensa, y fracturas patológicas que requieren inmovilización y, por ello, agravan el proceso. Nuestro paciente presenta deformidades articulares no sólo por la osteólisis carpotarsal y la sinostosis interfalángica, sino por las fracturas óseas que ha sufrido y el aplastamiento vertebral secundarios a su intensa osteoporosis.
Los resultados analíticos fueron normales, de acuerdo con los casos descritos2-4,6,7, y tanto la exploración neurológica como el estudio electromiográfico descartaron afección neuromuscular.
Nuestro diagnóstico de sospecha es el SW, descrito en 1969. Se caracteriza por osteólisis multicéntrica de predominio carpotarsal e interfalángico, grave osteoporosis generalizada y contracturas articulares progresivas, a las que pueden asociarse una facies tosca ("de carpa") con hipertrofia gingival y alteraciones cutáneas y corneales. La tosquedad de la cara ha llevado en algunos casos a realizar diagnóstico diferencial con la mucopolisacaridosis2,7, pero no resulta tan llamativa en nuestro paciente. Las alteraciones cutáneas descritas son hiperpigmentación, adelgazamiento cutáneo e hipertricosis, de las que nuestro paciente presentaba la última. En nuestro caso la alteración corneal principal es el queratocono, pero por el momento no se ha hallado las opacidades encontradas en otros pacientes con SW8.
Nuestro caso se diferencia del ST por presentar osteólisis y osteopenia severas, sin ensanchamiento de metatarso ni metacarpo. No ha desarrollado nódulos subcutáneos, que son característicos del ST y la primera y constante manifestación del síndrome de NAO, cuyas alteraciones esqueléticas son muy similares al SW.
Los cuadros de osteólisis son muy infrecuentes pero muy incapacitantes, por lo que se debe realizar estudio y tratamiento precoces. Al ser en su mayoría entidades hereditarias, los antecedentes familiares son muy relevantes para ofrecer un consejo genético adecuado.
