


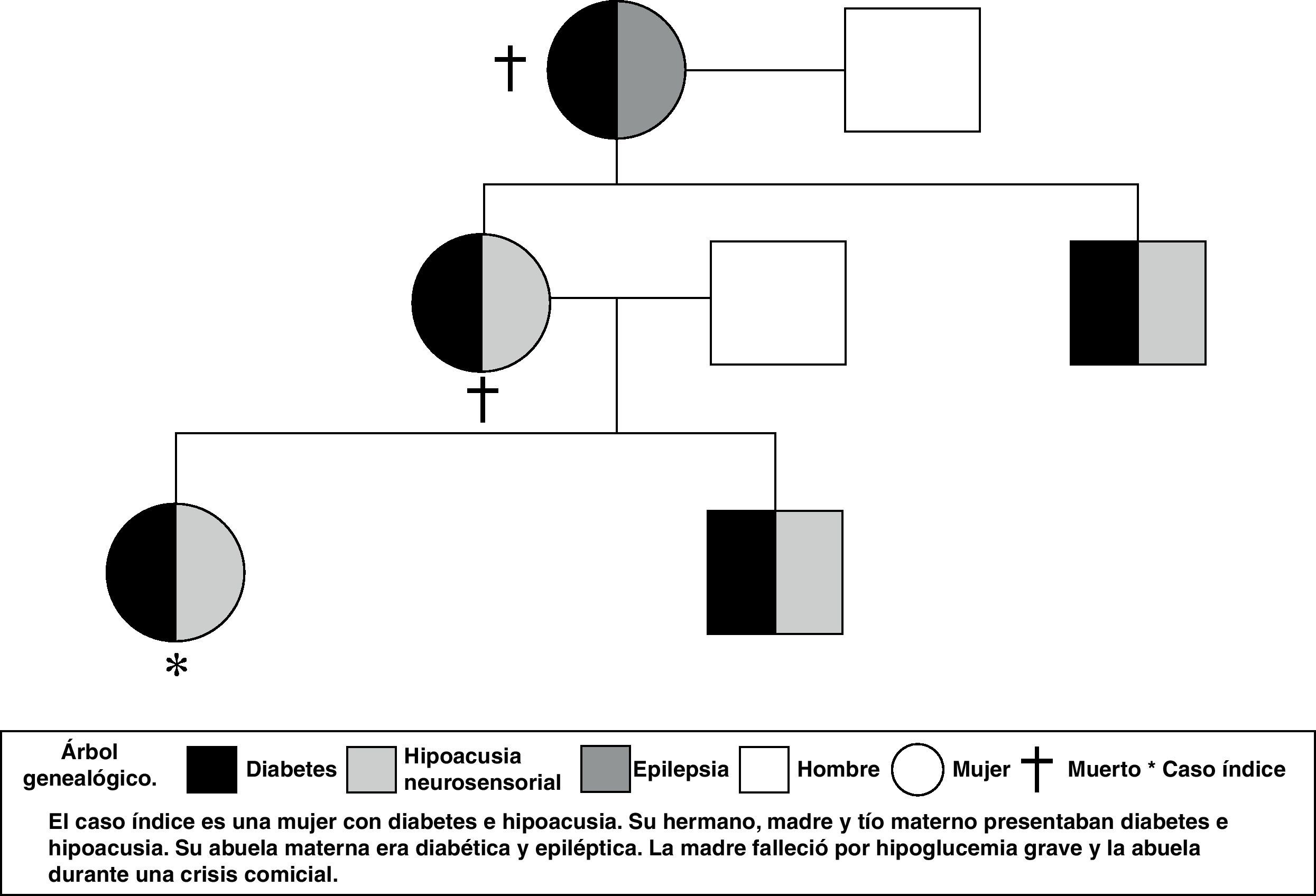
Presentamos el caso de una mujer de 34 años remitida para valoración de su diabetes mellitus (DM). Sus antecedentes personales eran: hipoacusia bilateral, otitis media crónica izquierda, pre-excitación anteroseptal asintomática con coronarias normales, y DM diagnosticada a los 24 años por hiperglucemia leve. Entre sus antecedentes familiares destacaban: abuela materna con DM y epilepsia; madre con DM desde los 21 años e hipoacusia; tío materno con DM e hipoacusia, y un hermano diabético diagnosticado a los 30 años también con hipoacusia. La madre había fallecido durante un episodio de hipoglucemia, y la abuela a los 36 años durante una crisis comicial (fig. 1).

Fue tratada inicialmente con hipoglucemiantes orales (metformina y sulfonilureas), y en los últimos 4 años con insulina, siempre con mal control glucémico. No refería complicaciones metabólicas agudas o crónicas. La exploración física fue normal con un índice de masa corporal de 23Kg/m2. En las exploraciones complementarias se objetivó: hemoglobina glucosilada 7,9%, péptido C 0,22ng/ml (valores de referencia: 1,00-4,00), y anticuerpos anti-descarboxilasa del ácido glutámico (GAD65), anti-citoplasma de células pancreáticas (ICA), anti-tirosina-fosfatasa (IA-2) y anti-insulina negativos. Teniendo en cuenta los antecedentes familiares y los datos de su DM, se realizó un estudio genético del ADN mitocondrial (mtDNA) en sangre (leucocitos). Empleando el método de secuenciación directa del gen tRNALeu (ácido ribonucleico transferente), se detectó la mutación 3243A>G en mtDNA con unos niveles aproximados de heteroplasmia del 31%. Por el método de PCR-RFLP (reacción en cadena de la polimerasa y polimorfismos del tamaño de los fragmentos de restricción mediante detección microfluída en Bionalyzer 2100 [Los Ángeles, EE.UU.]), la heteroplasmia fue del 30%. Se diagnosticó de diabetes e hipoacusia de herencia materna (MIDD), iniciándose tratamiento con coenzima Q10 150mg/día, y optimizando la terapia insulínica. No se realizó biopsia muscular para el estudio de la cadena respiratoria ni para la detección de fibras rojas rasgadas puesto que se trataba de una prueba invasiva y no aportaba datos adicionales en cuanto al diagnóstico y al tratamiento. Como comorbilidades asociadas al síndrome, se confirmó una hipoacusia perceptiva con umbral medio a 65 decibelios (dB) en el oído derecho y una hipoacusia mixta con umbral perceptivo medio a 65dB en el oído izquierdo. Presentaba un impedimento auditivo monoaural estimado derecho del 63%, monoaural estimado izquierdo del 95% y biaural estimado del 65%. El fondo de ojo y el ecocardiograma fueron normales, y la microalbuminuria normal.
Se dedujo que su hermano, madre, tío y abuela eran portadores de la mutación 3243A>G. Su madre y abuela habían fallecido por complicaciones asociadas a la misma. Se realizó consejo genético a la paciente y se recomendó estudio genético del hermano y el tío.
La diabetes mitocondrial1 representa entre el 0,2 y el 2% de todas las formas de diabetes. Su herencia es materna. La mutación causante más frecuente es la 3243A>G (sustitución de adenina por guanina en la posición 3243) del gen tRNALeu del mtDNA. Existen de 2 a 10 copias de mtDNA por célula (poliplasmia). En este tipo de síndrome coexisten copias de mtDNA mutado y no mutado en la misma célula (heteroplasmia). La proporción de mtDNA mutado y el tejido en que se encuentra condicionan el fenotipo. Si existen bajos niveles de heteroplasmia presentarán solo MIDD. A mayores niveles de heteroplasmia desarrollarán, además de lo anterior, el síndrome de MELAS (encefalomiopatía, epilepsia, acidosis láctica y episodios similares a ictus).
Las células con altas tasas de replicación tienden a seleccionar mitocondrias sanas (ej. leucocitos de sangre periférica), mientras que las células de baja tasa de replicación tienden a acumular mitocondrias anormales (por ejemplo, células musculares, células de la mucosa oral, y células del epitelio urinario). Es conveniente, por tanto, realizar el estudio de la mutación en estas últimas.
La diabetes mitocondrial tiene una penetrancia del 100%. La edad media de presentación es de 38 años. Mayores niveles de heteroplasmia condicionan un inicio más precoz. Se origina por una disminución de la secreción de insulina estimulada por la glucosa no mediada por mecanismo autoinmune2. La secuencia de acontecimientos3 sería: 1) la sustitución de adenina por guanina en la posición 3243 del mtDNA produce una dimerización del tRNA que ocasiona una alteración de la aminoacetilación; 2) aumento de la degradación mitocondrial de las proteínas codificadas por el mtDNA; 3) reducción de la actividad de los complejos enzimáticos de la cadena respiratoria; 4) reducción de la síntesis de adenosín trifosfato (ATP); 5) disminución del cociente adenosín trifosfato/adenosín difosfato (ATP/ADP), y 6) reducción de la secreción de insulina (a través de los canales de potasio dependientes de ATP) e hipotéticamente apoptosis de las células β. Puede ser confundida tanto con una DM tipo 1, como con una DM tipo 2. Siempre requerirán insulina, habitualmente tras una media de 3,9 años de evolución. Suelen desarrollar complicaciones microvasculares4 (retinopatía 8%, nefropatía 28%) y complicaciones macrovasculares4 (cardiopatía isquémica 7%, enfermedad vascular periférica 4%). El tratamiento consiste en secretagogos de la insulina y posteriormente insulina. Debe evitarse la metformina por el elevado riesgo de acidosis láctica que presentan estos pacientes. El consumo de hidratos de carbono debe ser apropiado puesto que las hipoglucemias desencadenan episodios similares a ictus.
La hipoacusia neurosensorial4,5 se presenta a una edad media de 33,2 años, antes, a la vez o después que la diabetes. Las estructuras del oído más afectadas son la estría vascular y las células ciliadas. Condiciona una pérdida de audición progresiva y casi universal. Hay una mayor progresión en varones y en aquellos con mayor nivel de heteroplasmia. El tratamiento consiste en el implante coclear.
La miopatía se manifiesta como dolor y debilidad en miembros inferiores, y fibras rojas rasgadas en la biopsia del músculo esquelético.
La epilepsia es debida a la hiperexcitabilidad originada por el decremento energético. Para su tratamiento se deben evitar aquellos fármacos que disminuyan los niveles de L-carnitina (benzodiacepinas, ácido valproico, fenitoína, fenobarbital) y emplear en su lugar propofol, si es necesario. Pueden aparecer episodios similares a ictus, también denominados «ictus metabólicos», por aumento del ácido láctico en la zona que ocasiona un descenso del pH y un aumento del flujo sanguíneo. En la arteriografía no se demuestra oclusión de los grandes vasos cerebrales y no presentan un límite territorial específico. Pueden ser desencadenados por hipoglucemia, fiebre o enfermedad intercurrente.
Pueden desarrollar proteinuria6 que se suele confundir con una nefropatía diabética. Histológicamente presentan: glomeruloesclerosis focal y segmentaria, con glomérulos hialinizados y necrosis de los miocitos de las arteriolas y arterias pequeñas.
Otras alteraciones posibles en este síndrome son: síndrome de pre-excitación, hipertrofia del ventrículo izquierdo, miocardiopatía dilatada7, distrofia macular retiniana, pseudo-oclusión intestinal, alteraciones neuropsiquiátricas, acidosis láctica y complicaciones durante el embarazo (prematuridad, placenta ácreta)8.
El diagnóstico se basa en la realización de estudios moleculares en los que se pretende buscar la mutación del mtDNA. Se pueden emplear dos técnicas: secuenciación y PCR-RFLP.
El tratamiento consiste en la administración de coenzima Q10, que es un transportador de electrones de la cadena respiratoria, y actúa como un antioxidante, protegiendo a los fosfolípidos de la membrana celular y al colesterol de las lipoproteínas de baja densidad del daño oxidativo causado por los radicales libres. Su administración, a dosis de 150mg/día, durante tres años retrasa la aparición de la insulinopenia y de la pérdida de audición y reduce los niveles de lactado post-ejercicio9. Otros autores no han corroborado estos hallazgos10. Se deben usar con precaución las estatinas, pues disminuyen los niveles de coenzima Q10. Existen tratamientos adicionales que se han empleado en otros síndromes mitocondriales: arginina, L-carnitina y complejos multivitamínicos.
En conclusión, aunque la prevalencia de la diabetes mitocondrial en la población diabética es baja, es importante diagnosticarla por su diferente pronóstico y tratamiento. Se debe sospechar cuando existe una historia personal y/o familiar de diabetes y sordera, complicaciones microvasculares que no se correlacionan con la duración de la diabetes, y en todo paciente diabético delgado con autoinmunidad pancreática negativa.
