


El panhipopitutarismo se define como el déficit completo de hormonas hipofisarias. La causa más frecuente en el adulto es el tumor hipofisario (61%), seguido de otras tumoraciones no hipofisarias como craneofaringiomas o meningiomas (9%)1. En función del tamaño tumoral y de la invasión de estructuras vecinas, a la clínica de déficit hormonal se suelen añadir otros síntomas, como alteraciones del campo visual si se comprime el quiasma óptico, o afectación de los pares craneales que transitan por el seno cavernoso. Entre las causas no tumorales, las más frecuentes son la silla turca vacía (7%) y el síndrome de Sheehan (6%)1.
Presentamos el caso de una paciente de edad avanzada con panhipopituitarismo y parálisis del iv par craneal.
Una mujer de 88 años ingresó en el servicio de medicina interna por cuadro de mal estado general, disnea a moderados esfuerzos y dolor torácico de características inespecíficas. La familia refería un deterioro progresivo del estado general desde hacía meses, con apatía, ánimo distímico, anorexia con escasa ingesta pero sin pérdida de peso, e intolerancia al frío. Entre sus antecedentes patológicos destacaban hipertensión arterial bien controlada, fibrilación auricular crónica, insuficiencia renal moderada y estreñimiento pertinaz de larga evolución. En la exploración física destacaba palidez y sequedad cutáneas con facies inexpresiva, bradipsiquia con lentitud en el curso del lenguaje, hiperdesviación y lateralización del ojo izquierdo en posición primaria de la mirada, tendencia a la hipotensión arterial (TA 100/60mm de Hg), tonos cardíacos apagados y arrítmicos e hipofonesis en ambas bases pulmonares. La campimetría por confrontación y el resto de la exploración neurológica, a excepción de la parálisis del IV par craneal izquierdo era normal.
La analítica de urgencias mostró anemia normocítica-normocrómica, deterioro de la función renal (creatinina 2,7mg/dl), hiponatremia (Na 126mEq/l) e hiperpotasemia (K 5mEq/l). El electrocardiograma mostraba fibrilación auricular con frecuencia ventricular media de 65 latidos por minuto y voltajes disminuidos. En la radiografía de tórax se observó cardiomegalia y derrame pleural bilateral de predominio izquierdo, por lo que se solicitó un TC tóraco-abdominal que confirmó el derrame pleural y un extenso derrame pericárdico con espesor mayor de 19mm. El ecocardiograma mostró aurícula izquierda dilatada con hipertrofia concéntrica grave del ventrículo izquierdo, función sistólica conservada y derrame pericárdico difuso sin compromiso hemodinámico.
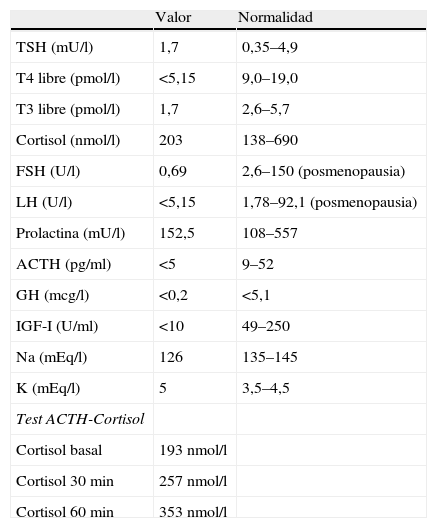
El perfil tiroideo puso en evidencia TSH de 1,7mU/l (0,35–4,9), T4 libre inferior a 5,15pmol/l (9,00–19,05) y T3 libre de 1,7pmol/l (2,6–5,7). Con estos resultados, y ante la sospecha de hipotiroidismo de origen central, se determinaron hormonas basales hipofisarias con los siguientes resultados: ACTH<5pg/ml (9–52), cortisol 203nmol/l (138–690), FSH 0,69U/l (2,6–150), LH<5,15 (1,78–92,1), prolactina 152,5mU/l (108-8-557,1), somatomedina C<10U/ml (49-250), GH<0,2μg/l (<5,1). Tras la administración de 250mcg de ACTH endovenoso, la concentración máxima de cortisol a los 60min fue de 353nmol/l (tabla 1).
Resultados de los valores analíticos y del test de ACTH-cortisol
| Valor | Normalidad | |
| TSH (mU/l) | 1,7 | 0,35–4,9 |
| T4 libre (pmol/l) | <5,15 | 9,0–19,0 |
| T3 libre (pmol/l) | 1,7 | 2,6–5,7 |
| Cortisol (nmol/l) | 203 | 138–690 |
| FSH (U/l) | 0,69 | 2,6–150 (posmenopausia) |
| LH (U/l) | <5,15 | 1,78–92,1 (posmenopausia) |
| Prolactina (mU/l) | 152,5 | 108–557 |
| ACTH (pg/ml) | <5 | 9–52 |
| GH (mcg/l) | <0,2 | <5,1 |
| IGF-I (U/ml) | <10 | 49–250 |
| Na (mEq/l) | 126 | 135–145 |
| K (mEq/l) | 5 | 3,5–4,5 |
| Test ACTH-Cortisol | ||
| Cortisol basal | 193nmol/l | |
| Cortisol 30min | 257nmol/l | |
| Cortisol 60min | 353nmol/l |
Con la orientación diagnóstica de panhipopituitarismo se inició tratamiento sustitutivo con glucocorticoides y posteriormente con levotiroxina, tras lo que la paciente presentó una clara mejoría clínica.
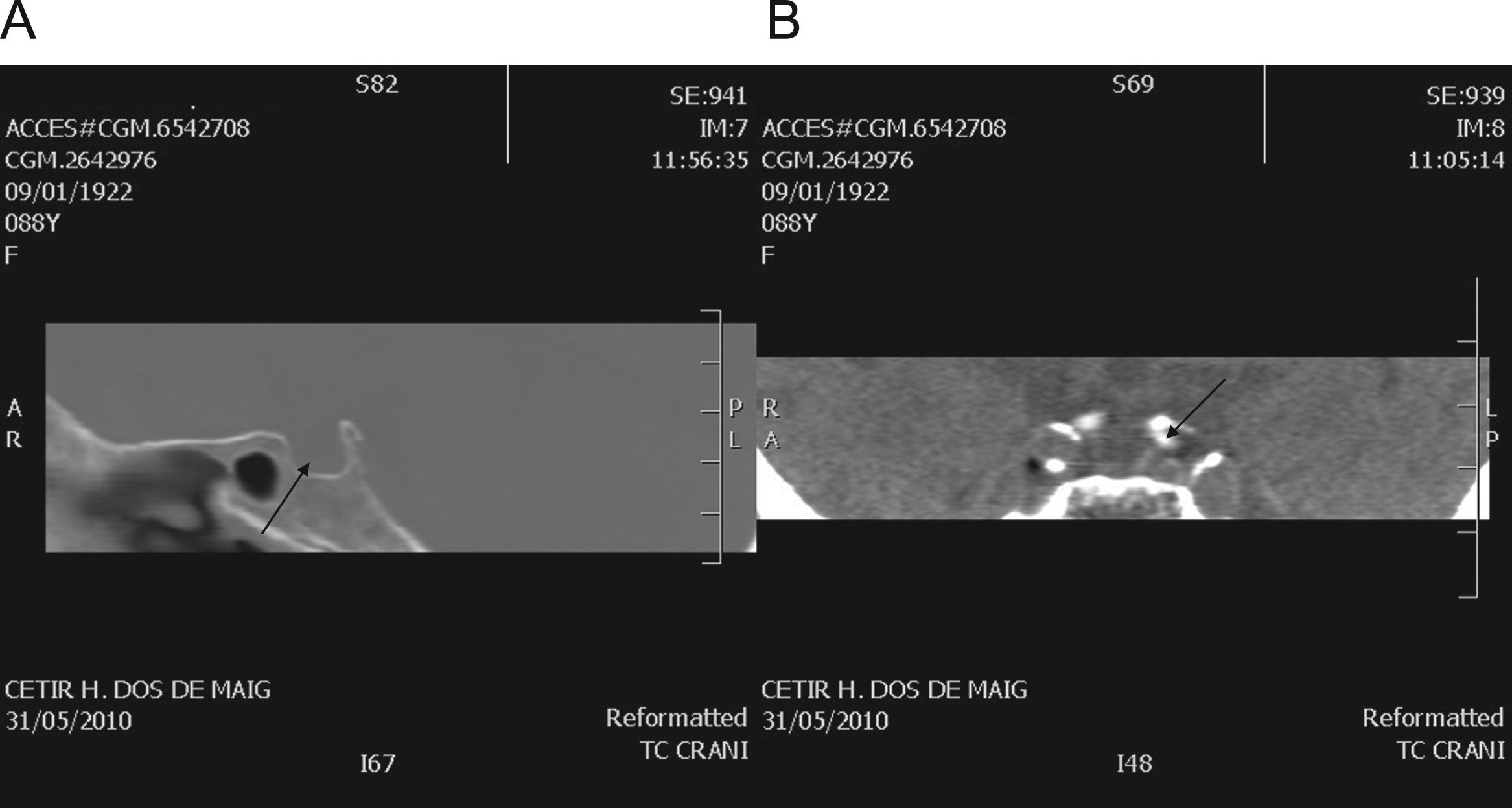
Para completar el estudio, se solicitó un TC centrado en hipófisis, al no poderse realizar una RM por claustrofobia, que mostró silla turca vacía (fig. 1A) y dilatación aneurismática de la arteria carótida interna a su paso por el seno cavernoso izquierdo, con impronta en el margen lateral del mismo (fig. 1B).
 Ocupación completa de la silla turca por líquido cefalorraquídeo (flecha). B) Dilatación de la arteria carótida interna con impronta a su paso por el margen lateral del seno cavernoso izquierdo (flecha).")
La silla turca vacía es una situación poco frecuente en pacientes jóvenes, pero su prevalencia aumenta con la edad, llegando en algunas series hasta el 48% en la octava década2,3, con un claro predominio entre mujeres. Uno de cada 4 pacientes que la presentan padecen déficits aislados de alguna hormona hipofisaria, especialmente de hormona de crecimiento, pero el panhipopituitarismo está presente únicamente en un 4,2–10,4% de los casos4. El diagnóstico suele llevarse a cabo tras la aparición de clínica secundaria a las alteraciones hormonales o bien de forma incidental. En un número reducido de casos son los síntomas secundarios a hipertensión intracraneal los que conducen al diagnóstico. Estas situaciones se han de tratar mediante derivaciones ventrículo-peritoneales, además de la sustitución hormonal de los ejes afectos.
Respecto a los aneurismas intracraneales, su prevalencia en la población general es del 1–6% según las series5–7. Se han descrito como causa de hipopituitarismo algunos casos en los que el aneurisma está localizado en la arteria comunicante anterior, aunque son excepcionales8. En la mayoría de ocasiones son asintomáticos, pero pueden ser potencialmente muy graves en caso de ruptura y hemorragia subaracnoidea.
En definitiva, la presencia de panhipopituitarismo diagnosticado en la edad adulta, junto a alteración de los nervios oculomotores, sugiere fuertemente la presencia de un macroadenoma hipofisario e invasión del seno cavernoso como diagnóstico más probable. Sin embargo, el caso aquí referido presentaba dos entidades independientes: silla turca vacía como causa de hipopituitarismo y dilatación aneurismática de la arteria carótida a su paso por el seno cavernoso como causa de parálisis del iv par craneal.
