


Se presenta el caso de un varón caucásico de 40 años de edad valorado en el servicio de urgencias por paraparesia de 1 h de evolución con hipopotasemia concomitante. Tras una progresión clínica brusca en las primeras 5 h de evolución, el cuadro se resolvió en relación con la normalización de la potasemia. La determinación analítica reveló un hipertiroidismo primario, y se estableció el diagnóstico de parálysis periódica tirotóxica. El abordaje terapéutico se centró en la administración de cloruro potásico, propranolol y metimazol. Pese a la aceptación generalizada del aporte potásico como primera medida terapéutica, la revisión de trabajos previos revela la necesidad de esclarecer la efectividad y el lugar del propranolol en el tratamiento de la crisis de parálisis periódica tirotóxica.
A 40-year-old Caucasian man presented to the emergency room of our hospital with bilateral lower extremity weakness with onset 1 hour previously and concurrent hypokalemia. After dramatic clinical progression for the first 5 hours, the episode resolved once serum potassium levels were normalized. Laboratory data revealed primary hyperthyroidism, indicating a diagnosis of thyrotoxic periodic paralysis (TPP). Treatment consisted of potassium, propranolol and methimazole administration. Although the mainstay of therapy is potassium replacement, the role of propranolol in improving the acute clinical manifestations of TPP has yet to be adequately clarified.
La parálisis periódica tirotóxica (PPT) es una complicación del hipertiroidismo, común en poblaciones asiáticas pero excepcional y, por lo tanto, difícil de reconocer cuando se presenta en España. Se han descrito episodios de PPT secundarios a estados hipertiroideos de cualquier origen, entre los que la enfermedad de Graves-Basedow figura como la causa más frecuente1. Generalmente se manifiesta en torno a la tercera década de vida y afecta llamativamente a los varones, con una razón de sexos de 70:1. La clínica endocrinológica puede preceder al episodio en varios meses o encontrarse el paciente previamente asintomático. La PPT puede ser la forma de presentación de la enfermedad tirotóxica2 y se caracteriza por episodios recurrentes de déficit motor agudo, limitado inicialmente a la musculatura proximal de las extremidades inferiores. En dos tercios de los casos se instauran durante el reposo nocturno, precipitados por la actividad física o la ingestión de hidratos de carbono alcohol en las horas previas3.
Ante un cuadro de estas características en un adulto joven, debe establecerse la sospecha de parálisis periódica hipopotasémica. La discriminación de un déficit real de potasio, con alteración del equilibrio acidobásico, de una redistribución de éste al compartimento intracelular, es fundamental en la orientación diagnóstica. El flujo masivo de potasio al interior de las células es la base fisiopatológica de las parálisis periódicas hipopotasémicas; en las formas familiares han sido definidas mutaciones en los genes que codifican canales de calcio (CACNA1S), sodio (SCN4A) y potasio (KCNE3). Sin embargo, la base genética de las formas esporádicas, con la PPT como forma más prevalente, continúa pendiente de dilucidar4. En un estudio5 se ha objetivado hipofosfatemia en el 80% de los casos de PPT. Las concentraciones séricas de fósforo en pacientes con PPT son inferiores a las de los pacientes con parálisis periódica hipopotasémica no tirotóxica6, lo que podría ser de utilidad en su diagnóstico diferencial.
Se presenta a continuación el caso de un varón caucásico de 40 años de edad, sin antecedentes personales ni familiares de enfermedad tiroidea, valorado en el servicio de urgencias por una paraparesia flácida de 1h de evolución. Se describen los hallazgos más significativos, así como el abordaje diagnóstico y terapéutico llevado a cabo. La fisiopatología de este fenómeno, sobre cuya base se realizará una discusión razonada de su tratamiento, es el objeto del último apartado.
CASO CLÍNICOVarón caucásico de 40 años de edad que una madrugada, horas después de ingerir una cantidad importante de hidratos de carbono, se despertó con dificultad en la movilización de las extremidades inferiores e incapacidad para mantener la bipedestación. En los últimos 6 meses había presentado de forma transitoria sensación de rigidez muscular y mialgia cuadricipital, con agravamiento y déficit motor proximal tras la realización de ejercicio físico. Refería un aumento del ritmo intestinal desde hacía 3 años y una pérdida de 3kg de peso. Sufría un estado continuo de nerviosismo, temblor distal, mala tolerancia al calor, sudoración y episodios de palpitaciones frecuentes.
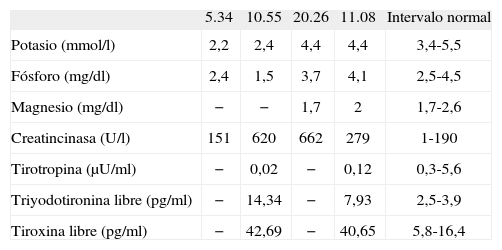
A su llegada al servicio de urgencias, la exploración física reveló una paraparesia flácida de predominio proximal, simétrica, con hiporreflexia osteotendinosa bilateral. La palpación muscular fue indolora y no se objetivó alteración en la sensibilidad. La palpación cervical mostró una glándula tiroides homogénea, de consistencia elástica, levemente aumentada de tamaño. El paciente presentaba taquicardia, una mínima retracción palpebral, piel caliente, sudoración y temblor distal en las extremidades superiores. La determinación analítica mostró hipopotasemia grave de origen no renal e hipofosfatemia leve sin alteración del equilibrio acidobásico (tabla 1). El electrocardiograma registró una taquicardia sinusal con un bloqueo auriculoventricular de primer grado y ondas U prominentes. En las horas siguientes, pese a la perfusión intravenosa de ClK a un ritmo de 5mEq/h, el cuadro progresó rápidamente hacia tetraparesia de predominio proximal y diplejía facial, por lo que se decidió el traslado a la unidad de cuidados intensivos y la administración intravenosa de inmunoglobulinas por la sospecha clínica de síndrome de Guillain-Barré (el estudio del líquido cefalorraquídeo, habiendo resultado normal, no descartaba esta posibilidad). Diez horas después de su llegada al servicio de urgencias, los resultados del perfil tiroideo revelaron hipertiroidismo primario. La enzima creatincinasa, en un principio normal, mostró entonces una elevación moderada, lo que reflejaba cierto grado de rabdomiólisis. Se mantuvo la perfusión de ClK y, con base en la sospecha clínica de PPT secundaria a hipertiroidismo primario autoinmunitario, se inició el tratamiento con metimazol 30mg/día, propranolol 20mg/8h y dexametasona 2mg/6h; 15h después de su inicio, el cuadro clínico se resolvió por completo coincidiendo con la normalización de la potasemia. Se suspendió el aporte de ClK y dexametasona, y se aumentó con fines profilácticos la dosis de propranolol a 40mg/8h, con buena tolerancia. La gammagrafía tiroidea con 99mTc-pertecnetato reveló un tiroides aumentado de tamaño en general, con captación homogénea. Los anticuerpos antitiroideos confirmaron el diagnóstico de enfermedad de Graves-Basedow (anticuerpos antitiroglobulínicos, 3.893 UI/ml; antiperoxidásicos, 5.726 UI/ml; antirreceptor de TSH, 152 mUI/ml). El estudio electrofisiológico (velocidad de conducción nerviosa y electromiograma) realizado 48h tras la resolución del episodio no reveló alteraciones significativas. Las alteraciones de la conducción y la repolarización cardíacas se normalizaron completamente. Al alta el paciente presentaba una mejoría parcial de la clínica adrenérgica, aunque todavía persistía clínica de mialgia y sensación de rigidez de la musculatura cuadricipital, sin déficit motor. Cinco meses después del episodio el paciente permanece asintomático y eutiroideo; está en tratamiento con propiltiouracilo, tras haber tenido reacción cutánea secundaria a metimazol y carbimazol. Se ha insistido en la conveniencia de un tratamiento radical, ya sea quirúrgico o radioablativo, puesto que únicamente un tercio de los pacientes con hipertiroidismo autoinmunitario tratados con antitiroideos de síntesis alcanzan la remisión a largo plazo, y el eutiroidismo es la única condición que garantiza que no recidivará una crisis de PPT3 potencialmente letal.
Parámetros bioquímicos en suero en las primeras horas de evolución
| 5.34 | 10.55 | 20.26 | 11.08 | Intervalo normal | |
| Potasio (mmol/l) | 2,2 | 2,4 | 4,4 | 4,4 | 3,4-5,5 |
| Fósforo (mg/dl) | 2,4 | 1,5 | 3,7 | 4,1 | 2,5-4,5 |
| Magnesio (mg/dl) | − | − | 1,7 | 2 | 1,7-2,6 |
| Creatincinasa (U/l) | 151 | 620 | 662 | 279 | 1-190 |
| Tirotropina (μU/ml) | − | 0,02 | − | 0,12 | 0,3-5,6 |
| Triyodotironina libre (pg/ml) | − | 14,34 | − | 7,93 | 2,5-3,9 |
| Tiroxina libre (pg/ml) | − | 42,69 | − | 40,65 | 5,8-16,4 |
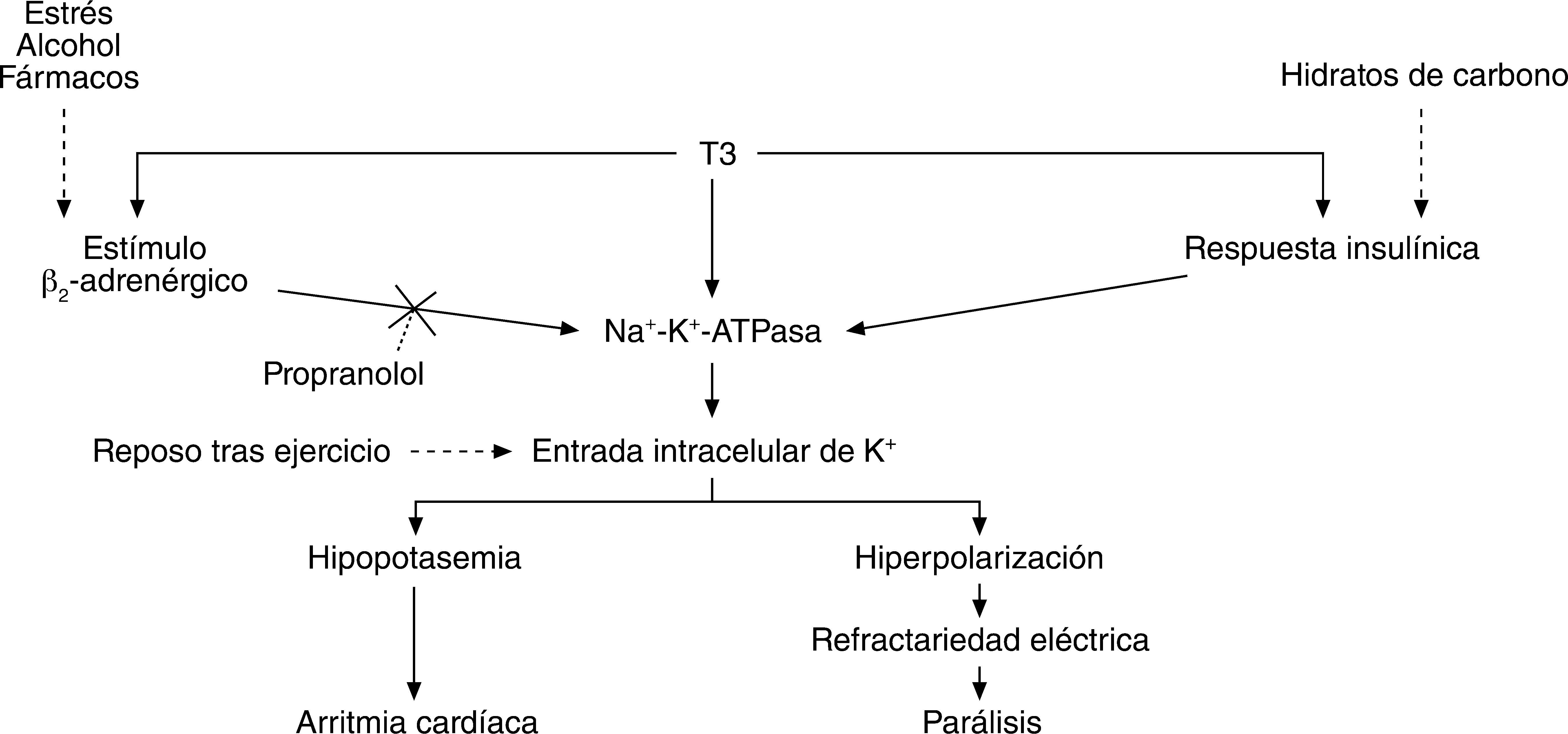
La patogenia de la PPT permanece mal conocida; la evidencia apunta a la existencia de una predisposición genética, revelada en la situación de hipertiroidismo, que favorece la activación anormal de la Na+-K+-ATPasa de la membrana de la fibra muscular7. El estado tirotóxico potencia la actividad de la Na+-K+-ATPasa por varias vías: directamente, incrementando el número de unidades de Na+-K+-ATPasa7, e indirectamente, aumentando el número y la sensibilidad de los receptores betaadrenérgicos y la respuesta insulínica. Los agonistas β2 activan la Na+-K+-ATPasa vía AMPc8 y la insulina induce la síntesis y la translocación de sus subunidades a la membrana celular9. Los distintos factores precipitantes de las crisis de PPT actuarían potenciando dicha actividad. La ingestión de hidratos de carbono intervendría mediante un aumento de la insulinemia, pues se ha demostrado que los pacientes con PPT tienen una respuesta insulínica exagerada respecto a pacientes hipertiroideos sin PPT10. La ingestión de alcohol, algunos fármacos y drogas (cafeína, anfetaminas, cocaína y agonistas β2-adrenérgicos), al igual que los episodios de estrés, actuarían por mediación adrenérgica4,11. El flujo masivo de potasio al medio intracelular conlleva dos hechos: la hipopotasemia y la hiperpolarización de la membrana de la fibra muscular, que adquiere un estado de refractariedad al estímulo nervioso (fig. 1).

La gravedad de las manifestaciones clínicas es proporcional al grado y la velocidad de instauración de la hipopotasemia12, por lo que de forma empírica se ha centrado el abordaje terapéutico en la administración de potasio con el objetivo de acelerar la resolución del episodio y prevenir la aparición de complicaciones cardiovasculares13. Sin embargo, se han descrito casos de hipopotasemia persistente pese a la perfusión de ClK11. Este hecho podría explicarse por la acción del propio potasio sobre la Na+-K+-ATPasa, directa e indirectamente mediante el incremento de la insulinemia, que perpetuaría de este modo su fuga al compartimento intracelular e impediría la normalización de la potasemia. Un estudio corrobora que, en efecto, el aporte a un ritmo de 10mEq/h podría suponer la resolución de la crisis en la mitad de tiempo que el grupo control, aunque permanezca desconocido el mecanismo de acción subyacente14. La eficacia de los suplementos de potasio en la profilaxis de las crisis no ha sido demostrada3.
Considerando el estímulo β2-adrenérgico como uno de los principales activadores de la Na+-K+-ATPasa, parece coherente que el uso de un antagonista betaadrenérgico no selectivo como el propranolol, al actuar sobre la base fisiopatológica del proceso, sea señalado como principal arma terapéutica y profiláctica de la PPT previa a la obtención del eutiroidismo. Pueden requerirse dosis elevadas (hasta 4mg/kg p.o.) titulando la dosis necesaria mediante la monitorización de la frecuencia cardíaca (asumiendo paralelos los efectos β2 en el flujo de potasio y β1 en el cronotropismo cardíaco)13. Por último, debe tenerse en cuenta que el aporte de potasio cuando no hay un déficit real puede abocar a una hiperpotasemia de rebote con consecuencias letales, lo que hace imprescindible un control estrecho de la potasemia. En un estudio14, el 40% de los casos tratados con 10mEq/h presentaron cifras de potasio superiores a 5,5mmol/l. Este riesgo será mayor en los casos en que, ante respuestas pobres a la perfusión de ClK, se haya procedido a la administración de propranolol, pues éste permite la salida del potasio acumulado en el compartimento intracelular. El tratamiento con propranolol a las dosis descritas, sin aporte de potasio concomitante, puede resolver rápidamente la PPT sin producir hiperpotasemia de rebote15.
La administración de dexametasona tuvo como objeto la inhibición de la secreción glandular de tiroxina (T4) y su conversión periférica en triyodotironina (T3) (mediante la inhibición de la 5'monodesyodasa), considerando el efecto directo e indirecto de ésta en la hiperactivación de la Na+-K+-ATPasa. Ciertamente el beneficio esperable se sustenta en una base teórica y sin evidencia empírica que la respalde; por otro lado, dado que su acción mineralocorticoidea es mínima, no consideramos que ésta interfiriera significativamente en la recuperación de la hipopotasemia, y en nuestra opinión es una medida adecuada.
En conclusión, la importancia de reconocer precozmente esta urgencia endocrinológica reside tanto en sus complicaciones potencialmente letales como en las diferencias sustanciales en el manejo terapéutico respecto a las demás formas de parálisis hipopotasémicas. La perfusión de ClK, considerado tratamiento de elección en numerosos trabajos, debería realizarse a un ritmo inferior a 10mEq/h y con monitorización, dado el elevado riesgo de hiperpotasemia de rebote. La inhibición no selectiva de los receptores betaadrenérgicos es una elección fundamentada en la comprensión de la fisiopatología de la PPT, y es una medida efectiva en la resolución y la profilaxis de las crisis que no produce hiperpotasemia por sí misma. No obstante, dada la escasez de casos recogidos tratados con propranolol en monoterapia, deberían realizarse estudios controlados para determinar su efectividad en comparación con el aporte de potasio.
