


El síndrome de Cushing endógeno es una entidad muy rara; su incidencia es de 2 a 4 casos por millón de habitantes y año. Hay que tener en cuenta la subestimación de los causados por secreción ectópica de corticotropina. La enfermedad de Cushing es la causa más frecuente del síndrome, unas 5 o 6 veces más frecuente que el síndrome de Cushing de origen suprarrenal. Tiene una incidencia entre 1,2 y 2,4 casos por millón de habitantes y año. La mujer tiene una frecuencia de 3 a 8 veces mayor que el varón para desarrollarla, 3 veces mayor para padecer afección tumoral suprarrenal, y de 3 a 5 veces mayor para padecer un síndrome de Cushing por tumor suprarrenal. La edad de diagnóstico del síndrome de Cushing varía con la etiología. La gran mayoría de las enfermedades de Cushing se deben a un adenoma hipofisario, aunque a menudo éste no sea visible mediante técnicas de imagen disponibles. El síndrome de Cushing no dependiente de corticotropina ocurre en aproximadamente el 20% de los casos. La mayoría se debe a adenomas (10%) o carcinomas (8%) suprarrenales. La hiperplasia micronodular bilateral y la hiperplasia macronodular son entidades raras, menos de 10% del total de casos del síndrome no dependiente de corticotropina. Existen formas familiares y esporádicas. La forma familiar, o complejo de Carney, y la hiperplasia macronodular bilateral no dependiente de corticotropina, en la que hay un aumento considerable del tamaño de las glándulas suprarrenales. Los signos y síntomas del síndrome de Cushing resultan directamente de la exposición crónica a un exceso de glucocorticoides. La mayoría de los síntomas y signos son muy prevalentes en la población general (hipertensión arterial, obesidad central, diabetes mellitus o intolerancia a los hidratos de carbono, osteoporosis y cambios fenotípicos característicos).
Endogenous Cushing's syndrome is a very rare entity, with an incidence of 2-4 cases per million inhabitants per year. Cases caused by ectopic ACTH secretion are under-diagnosed. Cushing's disease is the most frequent cause of endogenous Cushing's syndrome, which is 5 or 6 times more frequent than adrenal Cushing's syndrome, with an incidence of between 1.2 and 2.4 cases per million inhabitants per year.
Cushing's disease is 3-8 times higher in women than in men. The frequency of adrenal tumors is 3 times higher in women, while that of Cushing's syndrome due to adrenal tumors is 3-5 times higher. Age at diagnosis of Cushing's syndrome varies according to the etiology. Most cases of Cushing's disease are due to a pituitary adenoma, although the tumor may not be visible on the available imaging techniques.
ACTH-independent Cushing's syndrome is found in 20% of cases and is most frequently due to adenomas (10%) or adrenal carcinomas (8). Bilateral micronodular hyperplasia and macronodular hyperplasia are infrequent entities, representing less than 10% of all cases of ACTH-independent Cushing's syndrome. Both familial and sporadic forms exist: the familial form, or Carney complex, and ACTH-independent bilateral macronodular hyperplasia, in which the size of the adrenal glands is considerably enlarged.
The signs and symptoms of Cushing's syndrome are a direct result of long-term exposure to excessive glucocorticoids. Most signs and symptoms are highly prevalent in the general population (hypertension, central obesity, diabetes mellitus or carbohydrate intolerance, osteoporosis, and characteristic phenotypical alterations).
La verdadera incidencia del síndrome de Cushing es desconocida. Hay una gran subestimación en el registro de casos de Cushing iatrogénico, que es la causa más frecuente de síndrome de Cushing, teniendo en cuenta la prescripción frecuente de glucocorticoides.
El síndrome de Cushing endógeno es una entidad muy rara. Su incidencia es de 2-4 casos por millón de habitantes y año. No obstante, hay que tener en cuenta la probable infravaloración de los causados por secreción ectópica de corticotropina (ACTH). Teniendo en cuenta que la incidencia del carcinoma microcítico de pulmón (causante del 50% de los síndromes de Cushing ectópicos) es de 33.000 casos por millón de habitantes/año, y que al menos el 1% de estos tumores producen ACTH1-3, la incidencia de síndrome de secreción ectópica de ACTH se podría estimar en 660 por millón de habitantes y año. Sin embargo, la casuística conocida es inferior. La explicación radica en la agresividad del tumor, que enmascara el cuadro clínico del síndrome de Cushing. En otras ocasiones, el tumor produce precursores de ACTH (big-ACTH) cuya actividad biológica es muy escasa, aunque sean detectados por las técnicas de medición de ACTH.
La aparición de masas suprarrenales en series de autopsia oscila entre el 1,3 y el 8,7%, si bien la mayoría de éstas son hallazgos incidentales. De entre las masas suprarrenales productoras de cortisol, la mitad de los casos se debe a adenoma en la mayoría de las series. La incidencia de síndrome de Cushing por carcinoma suprarrenal es desconocida, pero se estima en 2 casos por millón de habitantes y año según el National Cancer Institute4.
La enfermedad de Cushing es la causa más frecuente de síndrome de Cushing endógeno, unas 5 o 6 veces más frecuente que el síndrome de Cushing de origen suprarrenal. Tiene una incidencia basada en estudios poblacionales entre 1,2 y 2,4 casos por millón de habitantes y año5,6. El único estudio sobre incidencia de la enfermedad de Cushing en España se ha realizado en la provincia de Vizcaya, con una prevalencia de 39,1 casos por millón de habitantes y una incidencia de nuevos casos de 2,4 casos por millón de habitantes y año6.
La distribución entre sexos varía según la etiología del síndrome de Cushing. Hace 25 años, el síndrome de Cushing ectópico ocurría con mayor frecuencia en el varón, con una relación 3:1. Sin embargo, el aumento del cáncer de pulmón relacionado con el tabaco en la mujer en las últimas décadas ha igualado esta relación. La mujer tiene una frecuencia 3-8 veces mayor que el varón para desarrollar una enfermedad de Cushing, 3 veces mayor para padecer enfermedad tumoral suprarrenal, y 3-5 veces mayor para padecer un síndrome de Cushing por un tumor suprarrenal.
La edad de diagnóstico del síndrome de Cushing también varía con la etiología. En el caso de tumores microcíticos de pulmón la edad corre paralela a la del tumor, con un aumento a partir de los 50 años. Los tumores carcinoides pueden aparecer a una edad más temprana, pero son muy raros en niños. La enfermedad de Cushing ocurre fundamentalmente en mujeres entre los 25 y los 45 años. Los tumores suprarrenales aparecen con una distribución bimodal, con un máximo en la primera década de la vida y otro a los 52 años para adenomas y 39 años para carcinomas suprarrenales.
En la edad infantil, la mitad de los casos de síndrome de Cushing se debe a carcinoma suprarrenal, mientras que la sexta parte se debe a enfermedad suprarrenal benigna. La enfermedad de Cushing acontece en la tercera parte, y son muy raros los tumores productores de ACTH ectópica.
En la mujer embarazada no es habitual encontrar síndrome de Cushing. Existen unos 70 casos publicados, y la mitad de ellos son síndrome de Cushing independiente de ACTH (el 42% adenoma, el 10% carcinoma), y el resto, sobre todo, debidos a una hiperplasia suprarrenal bilateral, la tercera parte de los cuales se atribuyó a enfermedad de Cushing. Sólo existen 3 casos publicados de producción ectópica de ACTH diagnosticados durante el embarazo.
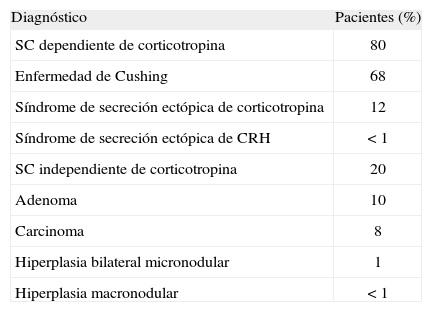
La prevalencia relativa de las distintas causas de síndrome de Cushing se muestra en la tabla 17. La enfermedad de Cushing causa la gran mayoría de los casos con síndrome de Cushing, seguido del síndrome de secreción ectópica de ACTH y síndrome de Cushing por enfermedad suprarrenal unilateral, a partes iguales.
Prevalencia relativa de las distintas causas de síndrome de Cushing (SC) endógeno*
| Diagnóstico | Pacientes (%) |
| SC dependiente de corticotropina | 80 |
| Enfermedad de Cushing | 68 |
| Síndrome de secreción ectópica de corticotropina | 12 |
| Síndrome de secreción ectópica de CRH | < 1 |
| SC independiente de corticotropina | 20 |
| Adenoma | 10 |
| Carcinoma | 8 |
| Hiperplasia bilateral micronodular | 1 |
| Hiperplasia macronodular | < 1 |
El síndrome de Cushing independiente de ACTH ocurre en aproximadamente el 20% de los casos. La mayoría se debe a adenomas (10%) o carcinomas (8%) suprarrenales. La hiperplasia micronodular bilateral y la hiperplasia macronodular son entidades raras, menos del 10% del total de casos de síndrome de Cushing independiente de ACTH. Los tumores productores de CRH ectópica son extremadamente raros (< 1%). En muchas ocasiones la secreción de CRH se realiza de forma combinada con la secreción ectópica de ACTH.
ETIOLOGÍAEnfermedad de CushingLa gran mayoría se debe a un adenoma hipofisario, aunque a menudo éste no es visible mediante técnicas de imagen disponibles. Estos tumores tienen sobreexpresados receptores tanto para CRH tipo 18 como para vasopresina tipo V39. La gran mayoría de los adenomas tienen un diámetro ≤ 1cm (microadenomas). Sólo el 5% son macroadenomas. Los macroadenomas tienden a producir mayores cantidades de ACTH y suprimirse menos con dexametasona10. La hiperplasia de células corticotropas como causa de enfermedad de Cushing es muy rara. En la enfermedad de Cushing hay una hipersecreción de ACTH hipofisaria con pérdida de su ritmo circadiano. El aumento de ACTH induce hiperplasia suprarrenal bilateral e hipersecreción de cortisol, con la consiguiente pérdida del ritmo circadiano de cortisol. Las células corticotropas de la hipófisis sana se encuentran atróficas debido a retroalimentación negativa de cortisol. También las células corticotropas tumorales mantienen cierta capacidad de retroalimentación negativa, disminuyendo la secreción de ACTH en respuesta a mayores aumentos de cortisol o glucocorticoides sintéticos (dexametasona). La diferencia entre células corticotropas normales y tumorales es sólo cuantitativa11. Según va aumentando la hiperplasia suprarrenal, la producción de cortisol en respuesta a una cantidad dada de ACTH va siendo cada vez mayor, lo que conduce a una disminución progresiva de la secreción de ACTH, que incluso puede llegar a un punto de "autosupresión". Este fenómeno se observa sobre todo en la hiperplasia suprarrenal macronodular grave12,13. En ella, las cifras de ACTH no superan los 15pg/ml, y se puede diagnosticar erróneamente síndrome de Cushing independiente de ACTH14. Sin embargo, no existe ningún caso bien documentado de progresión de hiperplasia macronodular hacia un síndrome de Cushing "completamente" independiente de ACTH.
Síndrome de secreción ectópica de ACTHEn este caso la hiperplasia e hiperfunción suprarrenal se deben a hipersecreción de ACTH por un tumor no hipofisario. El aumento de cortisol produce una inhibición fisiológica del CRH hipotalámico, así como de ACTH hipofisaria. Al contrario que en la enfermedad de Cushing, la hipersecreción de ACTH no se suprime con glucocorticoides, salvo en algunos casos habitualmente debidos a tumores carcinoides. Estos tumores pueden semejarse al fenotipo de la célula corticotropa y suprimirse con dexametasona y responder a CRH o a desmopresina15-17. En general, los tumores productores de ACTH tienden a secretar grandes cantidades de precursores de ACTH (big-ACTH), que pueden no detectarse con técnicas inmunorradiométricas, pero sí mediante radioinmunoanálisis. Un metaanálisis de 4 grandes series de síndrome de Cushing endógeno reveló una frecuencia de síndrome de Cushing ectópico de un 9-18%18. Aunque el carcinoma microcítico de pulmón es el tumor causal más frecuente de síndrome de Cushing ectópico en las series más antiguas19, la presencia de este tumor ha disminuido de manera sustancial en estudios más recientes18,20,21, desplazado por los tumores carcinoides. En una revisión publicada en 2002, de 530 casos de síndrome de Cushing ectópicos extraídos de 197 artículos, las 4 causas más frecuentes fueron el carcinoma microcítico de pulmón (27%), los carcinoides bronquiales (21%), los tumores insulares de páncreas (16%) y los carcinoides tímicos (10%)22. A pesar de que el carcinoma microcítico de pulmón sigue siendo el que con más frecuencia causa síndrome de Cushing ectópico, sólo un 1,4-4,5% de estos tumores se presentan con un cuadro clínico típico de hipercortisolismo1-3.
Síndrome de secreción ectópica de CRHEn este caso, extremadamente raro, la hipersecreción de CRH origina hiperplasia e hipersecreción de las células corticotropas hipofisarias, lo que resulta en aumento de ACTH, cortisol e hiperplasia suprarrenal bilateral23. En algunos casos hay supresión por dexametasona23. En otros casos hay secreción concomitante de ACTH y CRH por el tumor, sin supresión por dexametasona24-27.
Hiperfunción suprarrenal unilateral (adenoma/carcinoma suprarrenal). En todos los casos de hiperfunción suprarrenal primaria hay un aumento de la secreción de cortisol que conlleva la supresión de CRH y ACTH, con la consiguiente atrofia de las células corticotropas, así como de la zona reticular y fascicular de la corteza suprarrenal. La mayoría de los tumores suprarrenales son monoclonales, lo que indicaría mecanismos genéticos en su patogenia, tales como la activación de protooncogenes y la inactivación de genes supresores. En el caso del adenoma, la síntesis de cortisol es muy eficiente, por lo que los metabolitos intermedios y la DHEA-S se encuentran disminuidos en relación con el cortisol. En algunos adenomas suprarrenales se han encontrado receptores expresados de forma anómala capaces de estimular la secreción de cortisol, tales como GIP (péptido inhibidor gástrico)28-30, receptores betaadrenérgicos31, receptores de lutropina32, receptores de interleucina 133 y receptores de vasopresina V1a34-36. En el caso del carcinoma, la eficiencia para sintetizar cortisol a partir de colesterol es baja, y la producción de los metabolitos intermedios (p. ej., DHEA-S) es desproporcionadamente más elevada. Por ello, cuando aparece el cuadro clínico del hipercortisolismo, habitualmente el tamaño del tumor ya es muy grande. También se suelen encontrar elevados los precursores de la aldosterona, como desoxicorticosterona, 18 hidroxicorticosterona y corticosterona.
Hiperplasia bilateral micronodularExisten formas familiares y esporádicas. La forma familiar, o complejo de Carney, es una enfermedad autosómica dominante, caracterizada por dos hallazgos mayores: a) léntigos pigmentados y nevo azul de forma dispersa, y b) múltiples neoplasias, tanto endocrinas (tumor de células de Sertoli, tumores suprarrenales, hipofisarios o tiroideos) como no endocrinas (piel, mama, mixoma auricular, schwannomas)37,38.
Hiperplasia macronodular bilateral independiente de ACTHHay un considerable aumento del tamaño de las glándulas suprarrenales, que contienen múltiples verdaderos nódulos benignos, no pigmentados, > 5mm, separados por una estroma glandular hipertrófica. Patogénicamente, se ha intentado explicar por la sobreexpresión de receptores eutópicos, o inapropiada expresión de receptores ectópicos. En una serie de 20 pacientes con síndrome de Cushing de origen suprarrenal, todos los pacientes con hiperplasia macronodular bilateral (n = 6) expresaban receptores hormonales aberrantes, mientras que esto sólo ocurría en 3 de los 14 adenomas estudiados39. También se han encontrado respuestas de cortisol anormales frente a vasopresina, gonadotropina coriónica, lutropina o agonistas serotoninérgicos en 4 pacientes con síndrome Cushing subclínico e hiperplasia macronodular bilateral detectada de forma incidental40.
CUADRO CLÍNICOLos signos y síntomas del síndrome de Cushing resultan directamente de la exposición crónica a un exceso de glucocorticoides. La mayoría de los síntomas y signos son muy prevalentes en la población general (hipertensión arterial, obesidad central, diabetes o intolerancia a los hidratos de carbono). Por otro lado, ninguno de estos síntomas es suficientemente específico del síndrome, lo que en muchas ocasiones dificulta el diagnóstico. El diagnóstico precoz es importante debido la tasa elevada de comorbilidad asociada y mortalidad de los pacientes con síndrome de Cushing y a que la condición es tratable desde las fases inciales5.
La intensidad de los síntomas va a depender de factores como la duración y grado de hipercortisolismo, la asociación o no de hiperandrogenismo, y la causa del hipercortisolismo (hiperpigmentación en el caso de Cushing dependiente de ACTH). Los carcinomas suprarrenales y tumores productores de ACTH ectópica suelen producir síntomas relacionados con el propio tumor, enmascarando los efectos del hipercortisolismo (pérdida de peso en lugar de ganancia). Hasta el 10% de los incidentalomas suprarrenales presentan alguna alteración del eje hipotálamo-hipofiso-suprarrenal. Es el mal denominado síndrome de Cushing subclínico, debido a que no presentan las manifestaciones clínicas típicas del síndrome de Cushing. Sin embargo, estos pacientes presentan un perfil de riesgo cardiovascular mayor que la población normal, y similar a la de pacientes con síndrome de Cushing41,42.
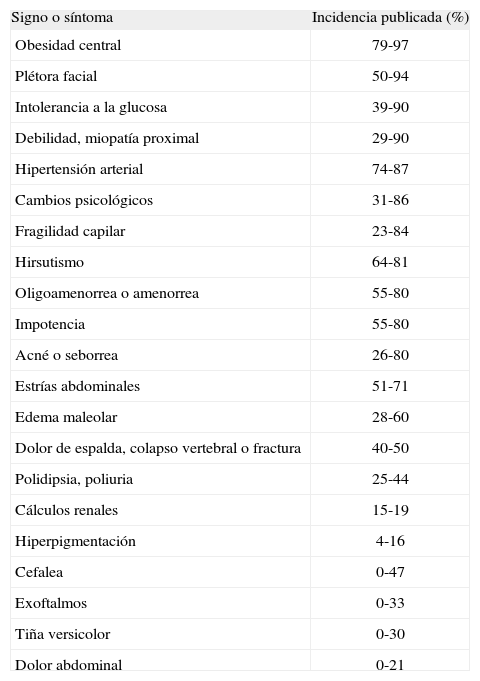
Los síntomas y signos del síndrome de Cushing se muestran en la tabla 227. Aunque ninguno de ellos es patonogmónico por sí mismo, la aparición de varios de ellos aumenta el índice de sospecha.
Signos y síntomas del síndrome de Cushing*
| Signo o síntoma | Incidencia publicada (%) |
| Obesidad central | 79-97 |
| Plétora facial | 50-94 |
| Intolerancia a la glucosa | 39-90 |
| Debilidad, miopatía proximal | 29-90 |
| Hipertensión arterial | 74-87 |
| Cambios psicológicos | 31-86 |
| Fragilidad capilar | 23-84 |
| Hirsutismo | 64-81 |
| Oligoamenorrea o amenorrea | 55-80 |
| Impotencia | 55-80 |
| Acné o seborrea | 26-80 |
| Estrías abdominales | 51-71 |
| Edema maleolar | 28-60 |
| Dolor de espalda, colapso vertebral o fractura | 40-50 |
| Polidipsia, poliuria | 25-44 |
| Cálculos renales | 15-19 |
| Hiperpigmentación | 4-16 |
| Cefalea | 0-47 |
| Exoftalmos | 0-33 |
| Tiña versicolor | 0-30 |
| Dolor abdominal | 0-21 |
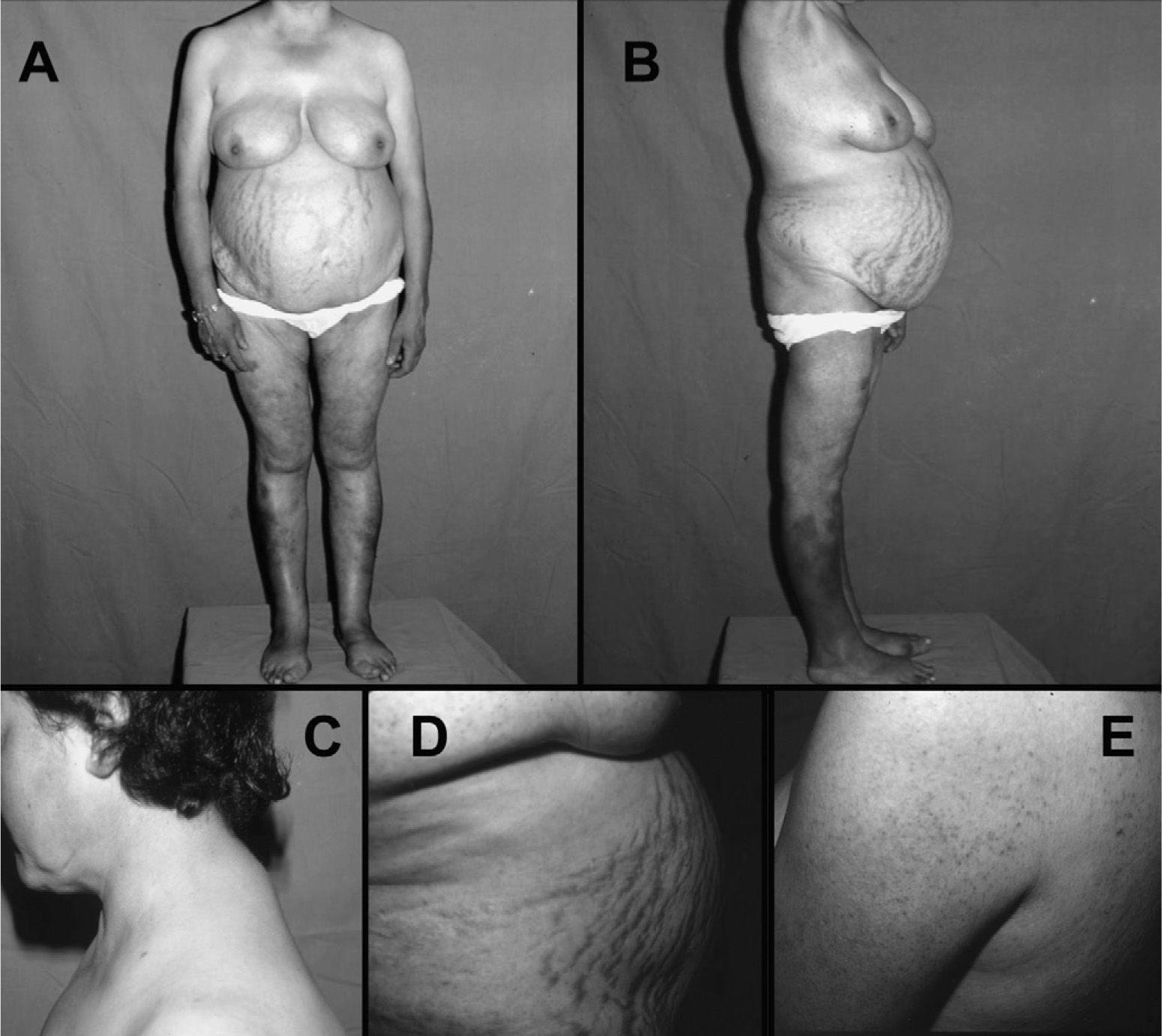
El síntoma más frecuente es la obesidad progresiva. Suele ser de distribución centrípeta afectando a cara, cuello y tronco (obesidad faciotroncular) (fig. 1). Suele respetar las extremidades, presentando en muchas ocasiones atrofia muscular. La obesidad generalizada no excluye el síndrome de Cushing, ya que puede estar presente de forma no infrecuente entre adultos con síndrome de Cushing43. En niños es típica la obesidad generalizada con retraso del crecimiento. La acumulación de grasa facial y cervicotorácica origina síntomas "típicos" como la "cara de luna llena", acompañado de plétora en mejillas, cara anterior del cuello y zona torácica fotoexpuesta. La "joroba de búfalo" (depósito dorsocervical) y pérdida del hueco supraclavicular confieren al paciente el aspecto de cuello gordo y corto (fig. 1). El depósito de grasa retroocular puede originar exoftalmos.
, la acumulación de grasa supraclavicular y \"giba de búfalo\" (C), la presencia de estrías abdominales vinosas gruesas (D) y foliculitis cutánea en espalda y brazos (E).")
Paciente con síndrome de Cushing por adenoma suprarrenal. Nótese la obesidad troncular con atrofia muscular proximal (A y B), la acumulación de grasa supraclavicular y "giba de búfalo" (C), la presencia de estrías abdominales vinosas gruesas (D) y foliculitis cutánea en espalda y brazos (E).
La debilidad muscular es muy frecuente. Se asocia a atrofia muscular proximal (glúteo o cintura escapular). Es típica la necesidad de ayuda de los brazos para incorporarse desde la posición de cuclillas, y en casos más graves dificultad para subir escaleras o incorporarse desde la posición de sentado. En casos de hipercortisolismo grave, la hipopotasemia resultante puede agravar aún más la debilidad muscular.
Las manifestaciones cutáneas del síndrome de Cushing se derivan de la atrofia cutánea, con adelgazamiento del estrato córneo y pérdida de la grasa subcutánea, dejando ver a trasluz los vasos sanguíneos subcutáneos. Existe una cicatrización lenta de las heridas menores y una tendencia a la dehiscencia de suturas de heridas quirúrgicas. La fragilidad capilar se debe a la pérdida de tejido celular subcutáneo. Hay tendencia a la aparición de múltiples hematomas ante mínimos traumatismos o en zonas de venopunción. A menudo es difícil mantener una vía venosa heparinizada. El estiramiento de la piel atrófica causa las típicas estrías vinosas, que característicamente son gruesas (> 1cm son muy sospechosas) y de color púrpura debido a la traslucidez la sangre venosa que circula por la dermis (fig. 1). Podemos observar también infecciones micóticas como la tiña versicolor en tronco y, más rara vez, onicomicosis. Es infrecuente la aparición de candidiasis oral. La hiperpigmentación secundaria al aumento de ACTH ocurre en los tumores productores de ACTH ectópica, y rara vez en la enfermedad de Cushing. El grado de hiperpigmentación dependerá de la duración y la magnitud de ACTH secretada. Se observa sobretodo en zonas fotoexpuestas (cara, cuello, dorso de las manos) y zonas de máxima fricción (codos, rodillas, cintura).
Las irregularidades menstruales ocurren hasta en el 80% de las mujeres con síndrome de Cushing. El síntoma más frecuente es la amenorrea/oligoamenorrea. Se ha encontrado una correlación entre el aumento de cortisol y disminución estradiol, pero no con el nivel de andrógenos44. La explicación podría ser la supresión de gonadorrelina mediada por el hipercortisolismo.
Las mujeres con síndrome de Cushing a menudo tienen signos de hiperandrogenismo45. Éstos suelen ser leves en la enfermedad de Cushing, y muy graves en el carcinoma suprarrenal, debido a las grandes cantidades de precursores androgénicos secretados por el tumor. Por el contrario, el hiperandrogenismo no ocurre en mujeres con el síndrome de Cushing por adenoma suprarrenal. El hirsutismo (leve y no generalizado), piel seborreica y acné faciotroncular (fig. 1), disminución de la libido y en ocasiones signos de virilización (en casos extremos, sobre todo carcinomas suprarrenales) son los síntomas habituales derivados del hiperandrogenismo. En varones es frecuente la impotencia y disminución de la libido.
La osteoporosis es muy frecuente en los pacientes con síndrome de Cushing. Se debe a una disminución en la absorción intestinal de calcio, a un aumento de la reabsorción y disminución de la formación ósea, y a una disminución de la reabsorción renal de calcio. Las fracturas por compresión vertebral o fracturas patológicas costales o de huesos largos son frecuentes. La necrosis avascular de la cabeza femoral ocurre en pacientes en tratamiento esteroideo en dosis altas, y es excepcional en los pacientes con síndrome Cushing endógeno. El aumento en la reabsorción ósea se puede acompañar de hipercalciuria y litiasis renal, y es muy rara la hipercalcemia.
La intolerancia a la glucosa también es frecuente en el síndrome de Cushing. Se atribuye al aumento de la neoglucogénesis inducida por cortisol y a la resistencia insulínica atribuida a la obesidad abdominal. La hiperglucemia franca ocurre en el 10-15%, especialmente en pacientes con antecedentes familiares de diabetes mellitus tipo 2. Aunque comúnmente se ha asumido que el síndrome de Cushing es una causa extremadamente rara de diabetes mellitus mal controlada, recientemente se han realizado dos estudios que encuentran una prevalencia elevada de pacientes con síndrome de Cushing dentro de esta población. Leibowitz et al encuentran una prevalencia de síndrome Cushing del 3,3% al estudiar a 90 sujetos obesos con diabetes mellitus mal controlada46. Prevalencia similar encuentran Catargi et al (el 2% con síndrome de Cushing y el 3,5% con síndrome de Cushing subclínico) al estudiar a una población de 200 pacientes diabéticos obesos mal controlados47. Por lo tanto, la presencia de diabetes mellitus mal controlada en un paciente con obesidad aumenta considerablemente la probabilidad de que contraiga síndrome de Cushing, en contra de lo que clásicamente se postulaba.
La hipertensión arterial es frecuente en el síndrome de Cushing. Se podría explicar por varios factores: a) sensibilidad periférica aumentada a los agonistas betaadrenérgicos; b) aumento en la producción del sustrato de renina, y c) activación de los receptores del túbulo renal tipo 1 (mineralocorticoides) en los hipercortisolismos graves (producción ectópica de ACTH). En este caso, el gran exceso de cortisol no puede ser inactivado por el riñón, lo que resulta en activación de los receptores de mineralocorticoides.
El riesgo cardiovascular de los pacientes con síndrome de Cushing está aumentado y determina la gran morbimortalidad de estos pacientes. El 80% de 49 pacientes con síndrome de Cushing obtuvieron un elevado o muy elevado riesgo cardiovascular estimado acorde con las guías de la Sociedad Internacional de Hipertensión/OMS (el 85% eran hipertensos; el 47%, diabéticos y el 41%, obesos)48. Los pacientes con incidentaloma suprarrenal y síndrome de Cushing subclínico también tienen un aumento del riesgo cardiovascular, similar al de los pacientes con síndrome de Cushing41. Los parámetros de riesgo cardiovascular de los pacientes con enfermedad de Cushing mejoran después de alcanzar la curación, aunque no llegan a alcanzar valores normales. Esto se podría explicar por la obesidad central residual y resistencia insulínica49,50.
Los fenómenos tromboembólicos, tales como tromboflebitis y tromboembolias, también han sido descritos asociados al síndrome de Cushing. Probablemente se deban al aumento en el factor VIII y factor de Von Willebrand, así como a la disminución de la actividad fibrinolítica. Es necesaria la anticoagulación profiláctica con heparina después de cualquier cirugía en estos pacientes.
Un gran abanico de síntomas neuropsiquiátricos han sido asociados al síndrome de Cushing, tales como labilidad emocional, depresión, irritabilidad, ansiedad, ataques de pánico e incluso paranoia o brotes psicóticos. El insomnio es una característica casi constante, relacionada con las elevadas concentraciones de cortisol durante las horas de sueño y a la ausencia de ritmo de cortisol. La depresión ocurre en dos tercios de los pacientes con síndrome de Cushing, y en los casos más graves incluso lleva al suicidio.
En el síndrome de Cushing existe riesgo elevado de infecciones bacterianas y oportunistas, especialmente en aquellos con mayor grado de hipercortisolismo (ectópicos)51-59. La inhibición de la inmunidad y de la secreción de citocinas, mediada por glucocorticoides, y la atrofia tímica que acontece en estos pacientes parecen tener un papel importante.
