


El síndrome de DiGeorge afecta a uno de cada 4.000 a 9.700 nacidos vivos1,2. La alteración genética responsable se ha hallado en la deleción de la región 22q11.2, por lo que actualmente se engloba dentro del grupo de trastornos del síndrome de deleción 22q11. Principalmente se describen malformaciones cardíacas, inmunodeficiencia, hipocalcemia, malformaciones en paladar con insuficiencia velofaríngea, dificultad para el aprendizaje y retraso del desarrollo. Otras asociaciones menos frecuentes son las malformaciones genitourinarias, enfermedad psiquiátrica y alteraciones osteoarticulares3. Como acrónimo se utiliza CATCH 22 (Cardiac defects, Abnormal facial features, Thymic hypoplasia, Cleft palate and Hypocalcemia).
Presentamos el caso de una paciente de 30 años remitida a consulta de Endocrinología por bajo peso. Como antecedentes personales constan: ductus arterioso intervenido a los 5 meses de edad, temblor con rasgos atípicos mioclónicos, paladar ojival, fractura de ambas rótulas, escoliosis lumbar, hipercifosis e hiperlordosis, condromalacia rotuliana, fibromialgia reumática, fenómeno de Raynaud bifásico, anemia y trombopenia leve oscilante, endometriosis y adenoidectomía. Sin hábitos tóxicos. Trabaja en empresa para discapacitados. Como antecedentes familiares de interés existen casos de neoplasias malignas, no de sordera o ceguera precoz ni enfermedades hereditarias conocidas. En la anamnesis dirigida niega pérdidas rápidas de peso, cambio reciente en la ingesta o hábitos incorrectos de alimentación. Realizaba natación que ha interrumpido por debilidad. Niega síntomas gastrointestinales. Se encuentra asíntomática desde el punto de vista tiroideo y de función suprarrenal. Niega polidipsia. Oligomenorrea sin mejoría tras la intervención de endometriosis ni bajo anticonceptivo oral. La paciente muestra su preocupación por infecciones respiratorias frecuentes, con unas tres neumonías al año objetivadas por su médico de Atención Primaria, sin atragantamientos previos. Meses antes se le indicó valproato por parte de Neurología. Toma diariamente ibuprofeno por dolores osteoarticulares. Exploración física: peso de 43,5kg con talla de 163cm, IMC de 16kg/m2. Disminución de masa muscular con fuerza conservada. Tensión arterial en decúbito supino 100/60 mmHg y en posición sentada 105/60 mmHg. Pulso 84 lpm, rítmico. Facies alargada con microstomía. Sin paresia de oculomotores. No se palpa bocio. Sin hiperpigmentación cutánea. Auscultación cardiopulmonar normal. No se palpan masas abdominales. Sin edemas. En el estudio endocrinológico obtenemos resultados normales en cuanto a función tiroidea, eje hipofisoadrenal, metabolismo hidrocarbonado y catecolaminas-metanefrinas en orina, con función renal y hepática normales. La analítica no muestra alteraciones en los parámetros nutricionales habituales y oligoelementos. Sin embargo, encontramos hipocalcemia (8,0mg/dL, valores normales 8,6-10,2), sin hipoalbuminemia acompañante, fósforo normal y calcidiol disminuido (15 mcg/L, VN 19-57) sin elevación compensatoria de parathormona (28 ng/L, VN 12-72). El magnesio determinado simultáneamente también es normal. La diuresis total en 24 horas es menor de 1.000mL en varias ocasiones, por lo que tomamos el cociente calcio/creatinina en orina reciente. En los estudios analíticos practicados con anterioridad los valores de calcemia están casi siempre dentro del rango de normalidad, con una determinación de parathormona (tabla 1), proteínas y albúmina también normales. Se revisa la historia pediátrica confirmando las neumonías de repetición desde los 15 meses de edad. Además consta resultado de Mantoux positivo, cultivos positivos para Giardia lamblia, e infecciones víricas comunes de la infancia. En este momento se emiten los diagnósticos de sospecha de bajo peso sin causa endocrinológica evidenciada, déficit leve de vitamina D y posible síndrome tipo CATCH 22. Mediante tratamiento dietético personalizado se consigue una ganancia de peso adecuada. En el servicio de Genética se confirma la sospecha clínica, emitiendo el diagnóstico de “síndrome de deleción 22q11.2”. En la ecografía abdominal no se detectan malformaciones del sistema urinario. En la siguiente revisión presenta calcemia normal sin suplementación oral de calcio o vitamina D, y sin cambios en cuanto a su sintomatología. El estudio de inmunocompetencia queda a cargo de Hematología. Por nuestra parte programamos el seguimiento analítico del metabolismo fosfocálcico para instaurar tratamiento en función de sus niveles de calcio, fósforo, calcidiol, calcitriol y densidad mineral ósea.
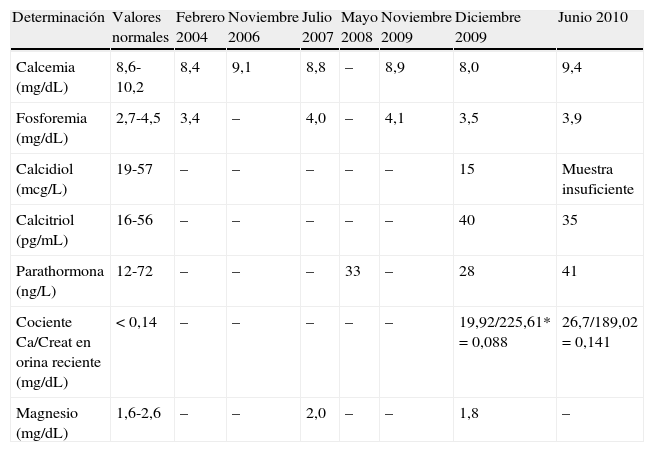
Evolución analítica de metabolismo fosfocálcico.
| Determinación | Valores normales | Febrero 2004 | Noviembre 2006 | Julio 2007 | Mayo 2008 | Noviembre 2009 | Diciembre 2009 | Junio 2010 |
| Calcemia (mg/dL) | 8,6-10,2 | 8,4 | 9,1 | 8,8 | – | 8,9 | 8,0 | 9,4 |
| Fosforemia (mg/dL) | 2,7-4,5 | 3,4 | – | 4,0 | – | 4,1 | 3,5 | 3,9 |
| Calcidiol (mcg/L) | 19-57 | – | – | – | – | – | 15 | Muestra insuficiente |
| Calcitriol (pg/mL) | 16-56 | – | – | – | – | – | 40 | 35 |
| Parathormona (ng/L) | 12-72 | – | – | – | 33 | – | 28 | 41 |
| Cociente Ca/Creat en orina reciente (mg/dL) | < 0,14 | – | – | – | – | – | 19,92/225,61* = 0,088 | 26,7/189,02 = 0,141 |
| Magnesio (mg/dL) | 1,6-2,6 | – | – | 2,0 | – | – | 1,8 | – |
–: determinación no solicitada; *: con presencia de piuria.
Resulta infrecuente la edad a la que la paciente fue diagnosticada. Los pacientes con síndrome de DiGeorge suelen diagnosticarse en edad pediátrica por cardiopatía congénita, hipocalcemia sintomática, infecciones recurrentes o no habituales, y/o fenotipo característico4. La paciente presentada tenía 30 años y desde el primer momento llamaba la atención las frecuentes neumonías que refería, así como el fenotipo típico y la intervención de ductus arterioso; sin embargo, no existía un diagnóstico establecido de hipocalcemia. Las infecciones respiratorias podrían haberse atribuido a problemas en la deglución dada su malformación facial. El resto de los antecedentes personales hacían suficientemente verosímil el cuadro sindrómico expuesto, o al menos nos obligaban a descartarlo.
En cuanto a la hipocalcemia, en ningún informe anterior o pediátrico se mencionaba una posible correlación con sus síntomas neurológicos. Además, su hipocalcemia no era constante en todos los estudios analíticos solicitados previamente, como puede ocurrir en casos de expresión parcial o atípica del síndrome. Hasta aquí se puede deducir que nos hallamos dentro del 40% de los pacientes sin alteración manifiesta del calcio. La paciente no describía claramente parestesias, su familiar negaba convulsiones en la infancia, y hasta el momento de nuestra valoración no se había emitido un diagnóstico definitivo de sus temblores por parte de Neurología. Las alteraciones neurológicas aparecen en un 8%, y se asume que la hipocalcemia es el agente causal en el 68% de los pacientes con crisis epilépticas3. Por tanto, nuestra paciente puede pertenecer a ese pequeño porcentaje de manifestaciones neurológicas con expresión atípica, en los momentos de hipocalcemia transitoria. También encontramos deficiencia de calcidiol en análisis recientes, había recibido tratamiento con valproato durante unas semanas y consultaba por bajo peso, por lo que podíamos haber pensado en un problema primariamente nutricional o de metabolismo de vitamina D. Sin embargo, al determinar la parathormona en presencia de hipocalcemia en ningún caso resultó elevada para compensar otra alteración, lo que llevó a la sospecha de una insuficiencia primaria de paratiroides al menos parcial.
Probablemente conforme aumenta la edad a la que se sospecha este cuadro sindrómico, más atípica es la expresión fenotípica, ya que los casos inequívocos desde el punto de vista clínico se han diagnosticado antes. Cabe pensar que los especialistas que atendieron previamente a nuestra paciente no se plantearon este diagnóstico por la presentación poco habitual que mostró en cuanto a hipocalcemia. Por ello, la sospecha debe provenir de pocos datos clásicos concordantes y con rasgos atípicos, tal y como ocurre en otro caso previamente publicado que se diagnosticó a los 15 años5. Con un diagnóstico precoz en nuestra paciente hay que suponer que se habría vigilado y tratado la hipocalcemia episódica, y se habrían atenuado las osteoartralgias debidas a sus malformaciones esqueléticas con un tratamiento rehabilitador.
Se ha observado suficiente heterogeneidad en la expresión fenotípica de pacientes con la deleción 22q11.26. Parece no existir un claro acuerdo en su nomenclatura, ya que hay múltiples denominaciones. Más bien se asume que son diferentes expresiones de la misma alteración7. Como clasificación funcional preferimos la que identifica por un lado a los pacientes con síndrome Velo-Cardio-Facial como aquellos que presentan principalmente alteraciones estructurales cardíacas con dismorfia facial, y por otro, los pacientes con síndrome de DiGeorge como aquellos con aplasia-hipoplasia tímica y paratiroidea. Por tanto, en nuestro caso se trataría de una forma intermedia entre ambos grupos.
