


El síndrome «morning glory» (SMG), aunque descrito inicialmente en 1929, debe su nombre a Kindler (1970)1. En él existe una displasia congénita del nervio óptico. Su incidencia es muy baja y está causado por un fallo en el cierre de la fisura embrionaria coroidea. Este síndrome, que tiene una herencia autosómica dominante, suele ser unilateral, aunque hay casos bilaterales, predomina en mujeres y se caracteriza por una excavación campaniforme, con tejido fibroglial central y unos vasos retiniales radiales que recuerdan a la flor dondiego de día o morning glory. El SMG frecuentemente se presenta con alteraciones oftalmológicas aisladas (disminución de la agudeza visual asociada a desprendimiento de retina en el 30 a 40% de los casos), pero se han descrito asociaciones sistémicas que incluyen alteraciones congénitas del prosencéfalo y de la línea media, con anomalías endocrinas progresivas, respiratorias o renales1,2.
Los encefaloceles basales (EB) congénitos se deben a un defecto óseo craneal y de la duramadre, con herniación extracraneal de estructuras intracraneales. Son anomalías raras, de difícil diagnóstico, con una incidencia estimada de 1:35.000 recién nacidos. Se clasifican en 4 tipos, de los cuales la variedad transesfenoidal es la menos frecuente (supone el 5% de los EB)3. Este tipo de encefalocele se debe a la persistencia del canal craneofaríngeo o transesfenoidal, con la herniación del tejido cerebral a través de él. Puede producirse por un defecto en el suelo de la silla turca, del esfenoides o del etmoides posterior; la variante menos frecuente es la transellar transesfenoidal. Con frecuencia se asocia a defectos de la línea media, con alteraciones hormonales hipotálomo-hipofisarias y del nervio óptico, entre ellas el SMG4. El SMG acompaña al 67% de los EB.
La incidencia de disfunciones hormonales en pacientes con EB es del 50-60%. La deficiencia de GH, hipogonadismo hipogonadotrópico, hipotiroidismo y diabetes insípida son los desórdenes más frecuentes. En una revisión de 15 casos con EB transesfenoidal, se evidenció que las deficiencias más frecuentes eran las de GH y hormona antidiurética (66,7 y 60%, respectivamente), seguidas por la deficiencia en gonadotropinas (33,3%), TSH (26,7%) y prolactina (13,3%)5.
El curso natural de la disfunción hipotálamo-hipofisaria es aún incierta, pero en la mayoría de los pacientes con EB se ha evidenciado disfunciones hormonales progresivas. Lamentablemente, hay muy pocos casos reportados con seguimiento endocrinológico en un periodo de tiempo superior o igual a 10 años3.
Presentamos un caso de SMG bilateral asociado a encefalocele transesfenoidal y panhipopituitarismo, en el cual los estudios de imagen y hormonales fueron fundamentales para el diagnóstico.
Una adolescente de sexo femenino de 166/12 años fue remitida por amenorrea y retraso del crecimiento, que había sido lento desde la infancia, pero sin estancamiento. A los 11 años inició axilarquia y pubarquia sin progresión. A los 16 años inició telarquia. No presentó menarquia. No presentaba poliuria ni polidipsia.
La paciente era producto de un embarazo controlado de 38 semanas, con parto eutócico, longitud de 49cm (+0,05DE) y un peso de 3.240g (+0,44DE). A los 3 meses de edad se le diagnosticó SMG bilateral, que le produjo a los 11 años desprendimiento de retina del ojo izquierdo y una visión reducida del ojo derecho. No tenía antecedentes de crisis comiciales ni otros datos de interés.
Exploración física a los 166/12: talla 145cm (−2,7DE), IMC 23,16Kg/m2 (+0,98DE), talla diana 163cm (−0,15DE). Edad ósea de 149/12 años. Microftalmia, nistagmo del ojo derecho, labio superior muy fino, paladar ojival. No existía bocio ni acúmulo de grasa en el abdomen. Estadio puberal 2 de Tanner. No existían otros datos significativos.
El estudio hormonal reveló TSH 1,67uUI/ml (VN 0,350-4,950), T4L 1,08ng/dl (VN 0,700-1,600), cortisol basal 21,20ug/dl (VN 5,00-25,00), ACTH 50,3pg/ml (VN 5-46). La respuesta del cortisol tras hipoglucemia insulínica fue normal (valor pico de cortisol 26,7ug/dl). PRL 61,6ng/ml (VN<20). GH basal<0,05ng/ml, con un pico de 0,06ng/ml (VN>7) en el test de clonidina no primado con estrógenos. IGF1 86ng/ml (VN 116-913). FSH basal 2,77 mUI/ml, LH basal 1,21mUI/ml. Estradiol 21pg/ml (VN impúber<12pg/ml). El test de GnRH muestra un pico de FSH 7,46mUI/ml y de LH 9,2mUI/ml. La densitometría ósea evidencia osteoporosis de −3,2DE (columna L1-L4). No se realizó osmolaridad urinaria debido a la ausencia de síntomas de diabetes insípida.
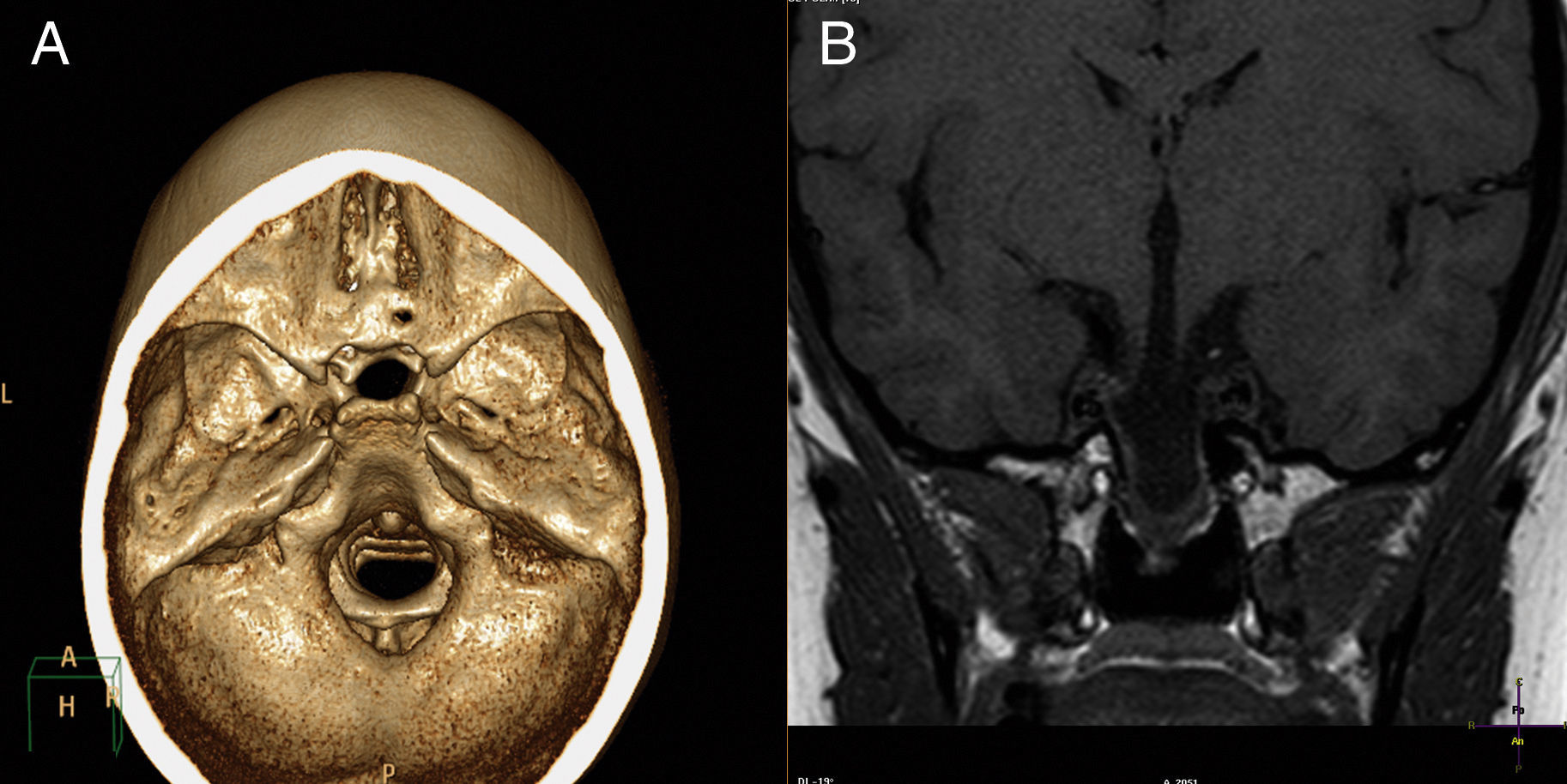
La TAC con reconstrucción 3D y la RMN cerebral (fig. 1) evidenciaron meningoencefalocistocele transesfenoidal, que contenía en su parte más inferior tejido hipotálamo-hipofisario displásico, agenesia del pico del cuerpo calloso y coloboma retiniano derecho. Se inició estrogenización con estrógenos transdérmicos6,7. No se administró GH dada la edad ósea, ya que este tratamiento es claramente efectivo si se administra a edades tempranas8.
 Tomografía axial computarizada tridimensional, donde apreciamos defecto óseo a nivel de base. B) Resonancia magnética nuclear. El corte coronal, en secuencia T1, muestra masa quística que se extiende a través de defecto óseo hacia nasofaringe")
Debido a la osteoporosis, además de la estrogenización se inició aporte de calcio y vitamina D. La hiperprolactinemia leve podía atribuirse a elongación del tallo hipofisario. No se realizó reparación quirúrgica del encefalocele, como está propugnado por muchos autores, ya que con frecuencia no es beneficioso por el riesgo de dañar el tejido funcionante9.
Nuestro caso presenta un SMG bilateral asociado a encefalocele transesfenoidal con alteraciones endocrinológicas: déficit de GH y de gonadotropinas con diagnóstico tardío de la afectación hipotálamo-hipofisaria, a pesar de conocerse el antecedente sindrómico. Por tanto, enfatizamos la necesidad de un estudio endocrinológico precoz y un seguimiento a largo plazo, debido a que las deficiencias hormonales pueden aparecer años después de realizado el diagnóstico inicial3,10. El estudio mediante TAC o RMN permite delimitar la anatomía de la masa herniada.
En resumen, ante el SMG es fundamental el estudio de imagen para descartar encefalocele, y la valoración de la función hipotálamo-hipofisaria para un diagnóstico y tratamiento precoz de los déficits hormonales. Las hormonas principalmente afectas son la GH, con repercusión en la talla final, y las gonadotropinas con la falta de estrogenización, que lleva, además de a una falta de desarrollo puberal, a una osteoporosis temprana. La terapia de reemplazo hormonal es muy efectiva6–8. Es fundamental recordar que las deficiencias hormonales pueden ser progresivas y manifestarse años después del diagnóstico inicial, siendo por ello necesario un adecuado seguimiento de la función del eje hipotálamo-hipofisario.
