


Se trata de una paciente de 16 años con antecedentes de pubertad precoz idiopática con imagen hipofisaria normal. Diez años después y en relación con el agravamiento de los episodios de cefalea de Horton, se realizó una nueva resonancia magnética craneal, en la que se objetivó una lesión sospechosa de sangrado intrahipofisario. Fue tratada con corticoides, que mejoraron la cefalea pero ocasionaron una complicación grave, la psicosis por esteroides.
La decisión de tratamiento quirúrgico y el estudio anatomopatológico de la lesión llevaron al diagnóstico diferencial entre craneofaringioma y xantogranuloma de silla turca. La evolución de la lesión (ausente 10 años antes), la indemnidad de la funcionalidad hormonal y visual, la resección total (muy difícil cuando se trata de craneofaringiomas) y la ausencia de recidiva hacen que el diagnóstico de xantogranuloma de silla turca sea prácticamente seguro.
We report the case of a 16-year-old girl with a history of idiopathic precocious puberty and normal results on pituitary imaging scan. Ten years later, a new cranial magnetic resonance imaging scan was performed due to worsening of episodes resembling Horton’s headache and a lesion suggestive of pituitary bleeding was detected. The headaches diminished with glucocorticoid administration but a severe complication, steroid psychosis, occurred.
Surgical treatment and pathological study of the lesion led to the differential diagnosis between craniopharyngyoma and xanthogranuloma of the sella turcica. The clinical progression of the tumor (not visualized 10 years previously), together with preservation of pituitary and visual function both before and after surgery, gross total removal of the tumor (difficult to achieve with craniopharyngioma) and the absence of recurrence provide strong support for the diagnosis of xanthogranuloma of the sella turcica.
Paciente que consultó por primera vez en nuestro servicio de endocrinología en 1994 a la edad de 6 años y medio por pubarquia precoz.
Entre sus antecedentes: peso al nacer, 2.850g; talla, 50cm; desarrollo psicomotor normal y enfermedades propias de la infancia. Como antecedentes familiares, su padre medía 172cm y presentó un desarrollo puberal en la edad normal; su madre medía 164cm, presentó la menarquia a los 14 años; tenía varias primas en rama materna con historia de menarquia precoz.
A la edad de 6 años apareció vello pubiano asociado a flujo vaginal y aumento de la velocidad de crecimiento. En la exploración física se trata de una niña normosómica con desarrollo armónico; talla, 118cm (percentil 60); talla en sedestación, 66,3cm; peso, 24kg; facies normal, sin manchas cutáneas; tiroides normal; vello pubiano incipiente, sin axilarquia ni telarquia. El resto de la exploración resultó normal.
Se realizó estudio en el que el hemograma y la bioquímica general, así como las pruebas de función tiroidea, resultaron normales. Estradiol plasmático, 18pg/ml; lutropina (LH) basal, 1,1mU/ml, que tras estímulo con LHRH ascendía a 13mU/ml; folitropina (FSH) basal, 1mU/ml que tras LHRH ascendía a 2,9mU/ml; 17 hidroxiprogesterona, 0,6ng/ml que tras estímulo con corticotropina (ACTH) ascendía a 1,8ng/ml. La ecografía abdominal y genital resultó normal. La edad ósea era de 8 años y 10 meses (pronóstico de talla, 151cm) y la resonancia magnética (RM) del área hipotalamohipofisaria no mostró ninguna alteración.
Con el diagnóstico de pubertad precoz idiopática, se inició tratamiento con triptorelina intramuscular cada 4 semanas, con buena respuesta; se mantuvo dicho tratamiento durante 30 meses. La paciente tuvo la menarquia a los 12 años, y su talla final fue 156cm y fue dada de alta.
En octubre del 2003, con 16 años, ingresó en nuestro servicio con sospecha de apoplejía hipofisaria. La paciente refería desde los 10 años episodios de cefalea punzante hemicraneal derecha de inicio progresivo a primera hora de la mañana asociada a ojo rojo y lagrimeo unilateral. Estos episodios se presentaban en racimos cada 2 o 3 meses y duraban 4–5h/día durante aproximadamente una semana. Había sido diagnosticada de cefalea de Horton o Cluster y por este motivo había seguido diferentes tratamientos. En los últimos meses la frecuencia de estos episodios había aumentado hasta hacerse prácticamente diarios. En la última semana la cefalea se había hecho más intensa, sin cambios claros en sus características, por lo que se realizó una RM craneal.
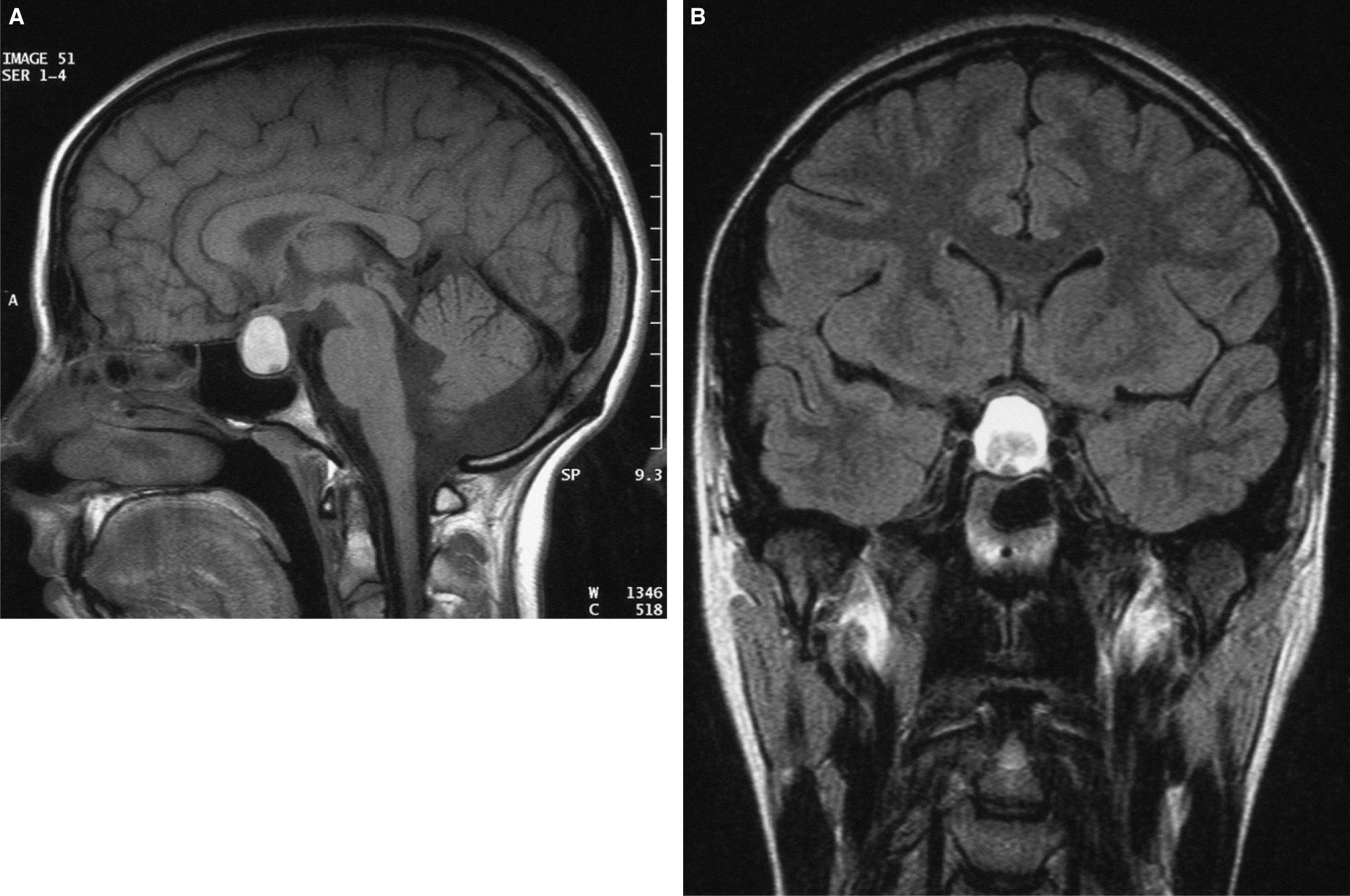
La RM mostraba una imagen de masa intrasellar bien definida, con una señal elevada en todas las secuencias compatible con macroadenoma hipofisario con signos de sangrado subagudo, desplazamiento del tallo, compresión y desviación quiasmática (fig. 1). Interrogada al respecto, no había notado alteraciones de la visión, mantenía reglas regulares y no presentaba otros datos clínicos de exceso o déficit hormonal. Se realizó valoración oftalmológica urgente: los movimientos oculares, reflejos pupilares, fondo de ojo y campimetría resultaron normales. Se obtuvieron muestras para determinaciones hormonales. Se valoraron en ese momento opciones de tratamiento: cirugía urgente frente a tratamiento con glucocorticoides, y se optó por manejo médico conservador. Se inició tratamiento con dexametasona inicialmente intravenosa (4mg/12h) y después vía oral, con lo que mejoraron de forma llamativa los síntomas de la paciente en las siguientes 24–48h.

El estudio hormonal hipofisario resultó normal.
Tras una semana de tratamiento y comprobar la ausencia de complicaciones oftalmológicas y estabilidad, la paciente fue dada de alta con dexametasona por vía oral.
Dos semanas después, la RM mostraba discreta mejoría radiológica con disminución de la afección quiasmática.
A las 4 semanas del alta la paciente fue llevada a urgencias por alteración del comportamiento y síndrome confusional agudo: la familia manifestaba que tras el alta la paciente presentó ánimo triste, que mejoró parcialmente tras iniciar tratamiento con sertralina. No presentó nuevos episodios de cefalea. Después comenzó a mostrarse progresivamente más inhibida, abandonó el aseo personal y expresó ideas alucinatorias.
A la exploración mostraba poca colaboración, estaba desorientada y bradipsíquica y contestaba con monosílabos. Los movimientos oculares eran normales y no presentaba otra focalidad neurológica. La bioquímica en sangre no mostró alteraciones, las determinaciones de tóxicos en orina (anfetaminas, cannabis, cocaína, benzodiacepinas, barbitúricos y opiáceos) fueron negativas. Se realizó una RM hipofisaria urgente, que no objetivó cambios con respecto a la previa, y un electroencefalograma urgente sin datos de actividad epileptógena. Descartada una causa orgánica, se solicitó valoración psiquiátrica y se le diagnosticó depresión y psicosis esteroidea, para lo que se la trató con haloperidol y se mantuvo la sertralina.
Se inició pauta descendente de glucocorticoides, con lo que reaparecieron los episodios de cefalea intensa. Por este motivo recomendamos tratamiento quirúrgico de la lesión hipofisaria. La clínica psiquiátrica mejoró progresivamente.
La paciente fue intervenida quirúrgicamente por vía transesfenoidal. Tras la apertura dural, se objetivó la salida de un líquido oleoso que indicaba craneofaringioma. El postoperatorio cursó sin complicaciones, salvo diabetes insípida transitoria.
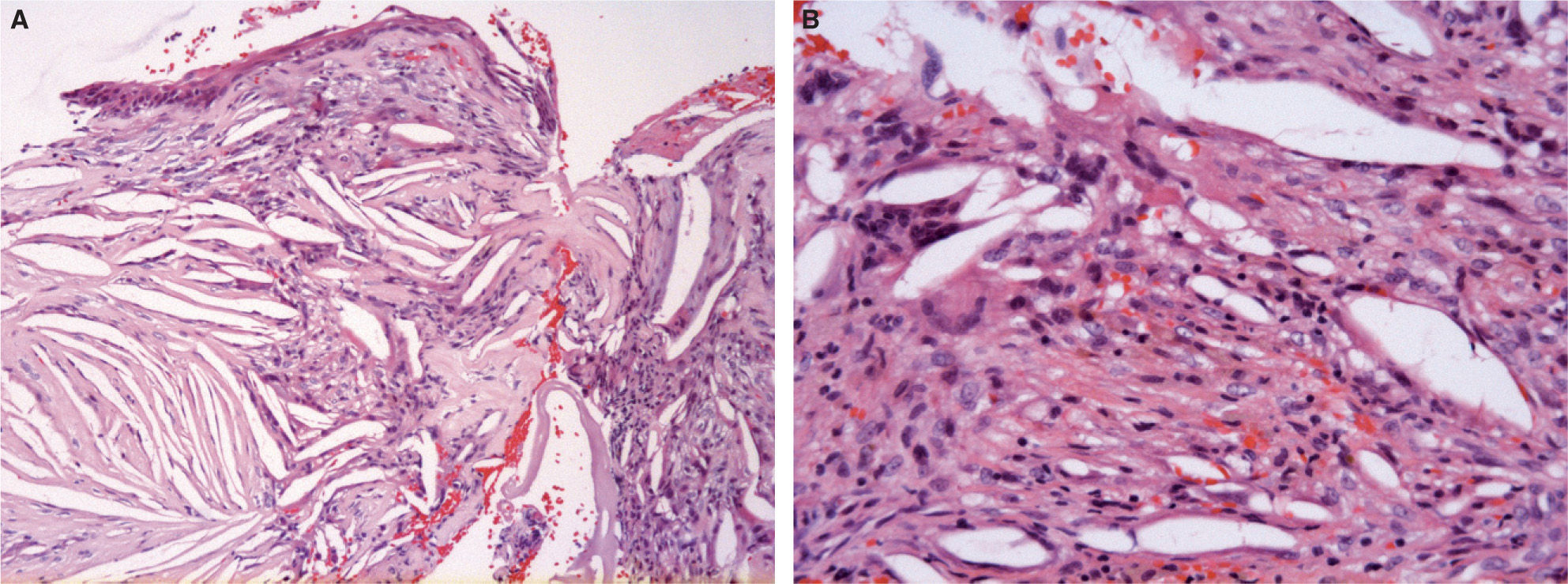
El estudio anatomopatológico de los fragmentos de tejido remitidos mostraba predominio de tejido xantogranulomatoso con abundantes cristales de colesterol entremezclados con detritos, celularidad inflamatoria, histiocitos, células gigantes de cuerpo extraño y algunos siderófagos. Se observó la presencia focal de epitelio escamoso aplanado sin queratinización. Todos estos datos eran compatibles con la impresión quirúrgica de craneofaringioma adamantinomatoso, pero el estudio histológico indicaba un xantogranuloma de silla turca (fig. 2).
 en el seno de tejido fibroso. Revestimiento parcial por epitelio escamoso (H-E, ×100). B: el infiltrado inflamatorio comprende células gigantes multinucleadas e histiocitos (H-E, ×200).")
El estudio hormonal posquirúrgico resultó completamente normal. La paciente quedó libre de cefaleas tras la cirugía y sólo ha tenido desde entonces dos episodios similares. Los controles de imagen realizados a los 6 meses y a los 2 años de la cirugía muestran cambios posquirúrgicos sin ningún dato de resto o recidiva tumoral. La paciente no ha sido radiada.
DISCUSIÓNLa pubertad precoz central o verdadera puede asociarse a multitud de lesiones en el sistema nervioso central, pero en la mayor parte de los niños no es posible identificar una causa, por lo que son diagnosticados de pubertad precoz idiopática.
La posibilidad de una neoplasia del sistema nervioso central obliga a realizar una tomografía o una RM craneal en cualquier paciente con una pubertad precoz central verdadera. El glioma óptico e hipotalámico, el astrocitoma, el ependimoma y rara vez el craneofaringioma pueden comprimir las vías neuronales que inhiben la generación de pulsos de LHRH en la infancia y causar una pubertad precoz1,2.
En nuestro caso, la prueba de imagen resultó (sin lugar a dudas) normal, la paciente fue diagnosticada de pubertad precoz idiopática y fue tratada, con buenos resultados.
A los 10 años de edad la paciente fue diagnosticada de cefalea en racimos en el servicio de neurología. Aunque la edad de comienzo de este tipo de cefalea es generalmente entre los 20 y los 50 años, puede ocurrir incluso en la primera década de la vida. Afecta más frecuentemente a varones. Se caracteriza por dolores de cabeza repetitivos que generalmente comienzan bruscamente en la zona periorbitaria y son siempre unilaterales. Se asocia a lagrimeo unilateral, ojo rojo, congestión nasal y náuseas. La patogenia no está totalmente aclarada; parece haber una alteración en neurotransmisión serotoninérgica, y el hipotálamo sería el lugar de activación3,4.
El aumento de intensidad de la cefalea y la disminución de los períodos de remisión llevaron a sus médicos a solicitar una RM craneal, que evidenció una masa intrasellar de aproximadamente 20mm de diámetro máximo que comprimía y desviaba el quiasma óptico y el tallo hipofisario. Por sus características radiológicas, la lesión era compatible con un macroadenoma hipofisario hemorrágico. Dado que no había alteración del nivel de conciencia ni oftalmoplejía y que la exploración oftalmológica era normal, se optó por tratamiento conservador con glucocorticoides, con lo que disminuyó discretamente el tamaño de la lesión y desapareció la cefalea5. La buena respuesta de la cefalea al tratamiento con glucocorticoides se ha descrito tanto en la cefalea en racimos como en la apoplejía hipofisaria.
La hipomanía y la depresión son efectos secundarios frecuentes en el tratamiento con glucocorticoides. Menos frecuentemente pueden aparecer cuadros psiquiátricos más graves como la psicosis. Se describe casi exclusivamente con dosis superiores a 20mg de prednisona y sobre todo en tratamientos largos. La respuesta a fármacos antipsicóticos es típicamente completa y ocurre a las 2 semanas de iniciar el tratamiento neuroléptico6. Nuestra paciente fue tratada con más haloperidol y sertralina, con mejoría en el plazo esperado; asimismo se disminuyó progresivamente los glucocorticoides, con reaparición de la cefalea, por lo que se optó por el tratamiento quirúrgico de la lesión intrasellar.
La anatomía patológica era compatible con craneofaringioma adamantinomatoso, pero apuntaba como más probable el diagnóstico de xantogranuloma de la silla turca.
Los craneofaringiomas son tumores derivados de remanentes embriológicos de la bolsa de Rathke que crecen lentamente desde el nacimiento hasta que son diagnosticados, generalmente en la infancia y la juventud, con otro pico de incidencia entre los 55 y los 65 años. Al diagnóstico pueden ser muy variables en sus características, desde tumores sólidos pequeños y circunscritos hasta grandes masas quísticas multilobuladas invasoras de más de 10cm de diámetro. Generalmente asientan en la zona del tallo hipofisario y el quiasma óptico y desde ahí pueden extenderse a estructuras circundantes. Con mucha frecuencia afectan a la silla turca, pero es raro que se manifiesten como masa exclusivamente intrasellar7.
La clínica depende de la edad. Los pacientes generalmente presentan hipofunción hipofisaria, alteraciones visuales y/o cefalea grave. En niños las manifestaciones hormonales más comunes son el déficit de hormona de crecimiento y el hipotiroidismo central, con el consiguiente retraso de crecimiento8. La pubertad precoz por esta causa es excepcional9,10.
La presencia de colesterol en una masa sellar puede ser el indicio principal del diagnóstico de craneofaringioma. Los espacios aciculares ópticamente vacíos (clefts) de colesterol son comunes en el craneofaringioma adamantinomatoso, aunque también pueden aparecer en los quistes de la bolsa de Rathke y en los adenomas hipofisarios.
Los xantogranulomas de la silla turca son granulomas de colesterol intrasellares y se ha señalado que se trata de una entidad clínica y patológicamente diferente del craneofaringioma adamantinomatoso11. Se diagnostican sobre todo en adolescentes y adultos jóvenes. En general son de tamaño más pequeño que los craneofaringiomas y de localización preferentemente intrasellar. Presentan calcificaciones con menor frecuencia y también es menos frecuente que cursen con alteraciones visuales. Se describe una mejor resecabilidad con un mayor porcentaje de resección total en la cirugía, y en general tienen un curso más favorable. En lo referente a su patogenia, es posible que puedan crecer como reacción a hemorragia, necrosis o inflamación12.
En el diagnóstico diferencial de este caso, debe mencionarse la hipofisitis xantomatosa. En esta entidad es característico, desde el punto de vista anatomopatológico, el gran predominio de histiocitos espumosos en el intersticio de una adenohipófisis relativamente conservada, con linfocitos y células plasmáticas; no se observa revestimiento epitelial, fibrosis, células gigantes multinucleadas, hendiduras de colesterol ni hemosiderina13.
Las características radiológicas de la lesión que presentaba nuestra paciente, con ausencia de calcificaciones, su pequeño tamaño y su localización exclusivamente intrasellar, junto con los datos clínicos con ausencia al diagnóstico de alteraciones visuales a pesar del contacto con el quiasma, y la indemnidad del funcionamiento hipofisario nos hacen pensar en el xantogranuloma de silla turca como diagnóstico más probable. Esto se apoya en el hecho de que 10 años antes la paciente tenía una RM hipofisaria completamente normal. Hasta donde hemos podido documentarnos, sólo ha sido publicado un caso de aparición de novo de un craneofaringioma en una paciente diagnosticada de esclerosis múltiple y con una RM 2 años antes sin lesiones hipotalamohipofisarias14.
La mayor parte de los craneofaringiomas son difíciles de resecar, e incluso tras la resección "completa" tienden a recurrir, por lo que algunos autores recomiendan la radioterapia como tratamiento adyuvante. La resección completa sin ninguna complicación perioperatoria, salvo diabetes insípida transitoria, y la ausencia de recurrencia tras 3 años de seguimiento también respaldan nuestro diagnóstico.
