


Obesity is associated to significant disturbances in endocrine function. Hyper insulinemia and insulin resistance are the best known changes in obesity, but their mechanisms and clinical significance are not clearly established. Adipose tissue is considered to be a hormone-secreting endocrine organ; and increased leptin secretion from the adipocyte, a satiety signal, is a well-established endocrine change in obesity. In obesity there is a decreased GH secretion. Impairment of somatotropic function in obesity is functional and may be reversed in certain circumstances. The pathophysiological mechanism responsible for low GH secretion in obesity is probably multifactorial. There are many data suggesting that a chronic state of somatostatin hypersecretion results in inhibition of GH release. Increased FFA levels, as well as a deficient ghrelin secretion, probably contribute to the impaired GH secretion. In women, abdominal obesity is associated to hyperandrogenism and low sex hormone-binding globulin levels. Obese men, particularly those with morbid obesity, have decreased testosterone and gonadotropin levels. Obesity is associated to an increased cortisol production rate, which is compensated for by a higher cortisol clearance, resulting in plasma free cortisol levels that do not change when body weight increases. Ghrelin is the only known circulating orexigenic factor, and has been found to be decreased in obese people. In obesity there is also a trend to increased TSH and free T3 levels.
La obesidad se asocia con importantes anomalías en la función endocrina. La hiper insulinemia y la resistencia a la insulina son las dos alteraciones mejor conocidas, aunque sus mecanismos y su significado clínico no están claros. El tejido adiposo se considera un órgano endocrino con secreción hormonal; el aumento en la secreción de leptina, una señal de saciedad, por el adipocito es una alteración característica. En la obesidad hay una disminución en la secreción de hormona de crecimiento; esta alteración en la función somatotropa de la obesidad es funcional y se puede revertir en determinadas circunstancias. El mecanismo fisiopatológico responsable de la hiposecreción de GH en la obesidad es probablemente multifactorial. Existen muchos datos que sugieren que un estado crónico de hipersecreción de somatostatina resulta en una inhibición de la liberación de GH; el aumento de los ácidos grasos libres probablemente contribuye a esta alteración, así como un déficit en la secreción de ghrelina. En mujeres, la obesidad abdominal se asocia a hiperandrogenismo y a niveles disminuidos de proteína transportadora de hormonas sexuales. Los hombres obesos tienen niveles de testosterona y concentraciones de gonadotropinas disminuidos, especialmente en los casos de obesidad mórbida. La obesidad se asocia con un aumento en la tasa de producción de cortisol, que se compensa con un aumento del aclaramiento del mismo, lo cual resulta en niveles plasmáticos de cortisol libre que no se modifican con el aumento del peso corporal. Ghrelina es el único factor orexígeno circulante conocido y se ha visto que se encuentra disminuido en humanos obesos. En la obesidad hay también una tendencia a aumentar las concentraciones de TSH y T3 libre.
Obesity is defined as excess body fat. Clinical management of obesity is complex and often provides poor results.1 Abdominal, or central, obesity reflects the amount of visceral fat and is directly related to insulin resistance and cardiovascular events.
There is an epidemic of overweight and obesity worldwide. There is a growing prevalence of excess weight throughout the United States, where approximately 65% of the adult population is overweight or obese.2 A comparison of the 1976–19803 and 1999–20002 periods shows a 40% increase in the prevalence of overweight (from 46% to 64.5%) and a 110% increase in the prevalence of obesity (from 14.5% to 30.5%). Spain is not free from this epidemic, and the prevalence of obesity and overweight in our country has dramatically increased.4 According to some reports, the prevalence rates of excess weight (obesity plus overweight) and obesity are approximately 65% and 25%, respectively.5
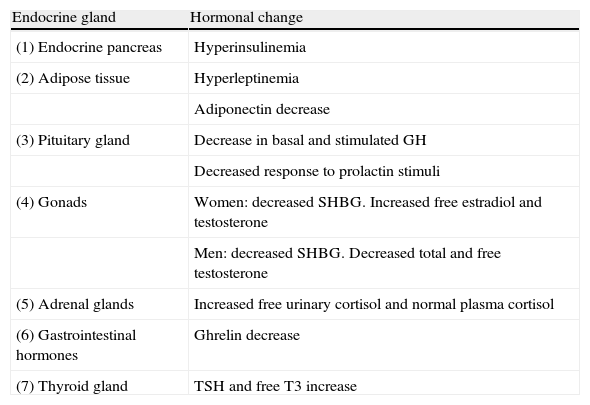
Like hypertension or diabetes, obesity is a chronic disease. The etiology of obesity is an imbalance between the energy obtained from intake and energy consumed. Excess energy is stored as body fat in adipocytes, which grow and/or increase in number. This adipocyte hypertrophy and hyperplasia is the pathological lesion characteristic of obesity. An increase in adipose tissue causes the clinical problems associated with obesity, due to either the weight of the extra fat mass or to the increased secretion of free fatty acids, many peptides, and other adipokines by hypertrophic adipocytes. Obesity and overweight are associated with a number of endocrine and metabolic changes (Table 1). It is thought that most changes are secondary to obesity, because they may be induced by overnutrition and are reversed by weight loss. Obesity is associated with changes in the plasma levels of certain hormones and changes in the secretion and/or clearance patterns. Some of these changes are secondary to obesity, while others could play a role in its pathogenesis.
Main endocrine changes in obesity.
| Endocrine gland | Hormonal change |
| (1) Endocrine pancreas | Hyperinsulinemia |
| (2) Adipose tissue | Hyperleptinemia |
| Adiponectin decrease | |
| (3) Pituitary gland | Decrease in basal and stimulated GH |
| Decreased response to prolactin stimuli | |
| (4) Gonads | Women: decreased SHBG. Increased free estradiol and testosterone |
| Men: decreased SHBG. Decreased total and free testosterone | |
| (5) Adrenal glands | Increased free urinary cortisol and normal plasma cortisol |
| (6) Gastrointestinal hormones | Ghrelin decrease |
| (7) Thyroid gland | TSH and free T3 increase |
The most characteristic endocrine change in obesity is increased insulin secretion. Obese people have increased insulin concentrations. Basal integrated 24-h insulin secretion is three to four-fold greater in obese subjects as compared to thin control subjects.6 Both obesity and type 2 diabetes mellitus are associated with insulin resistance,7 but most insulin-resistant obese subjects do not develop hyperglycemia. For obesity and insulin resistance to be associated with type 2 diabetes, the pancreatic beta cell must be unable to compensate for the decrease in insulin sensitivity.7
Adipose tissue modulates the metabolism by releasing free fatty acids and glycerol, hormones such as leptin, adiponectin, and proinflammatory cytokines such as TNF-alpha or interleukin-6. The release of non-esterified fatty acids is the most important single factor modulating insulin sensitivity. Increased levels of non-esterified fatty acids (NEFAs) are seen in obesity and type 2 diabetes, and are associated with the insulin resistance found in both.7
Body fat distribution is itself a determinant of insulin sensitivity.7 Slim individuals with peripheral fat distribution have a greater insulin sensitivity than slim people with central fat distribution. Intra-abdominal and subcutaneous fat are also different. Intra-abdominal fat is more lipolytic than subcutaneous fat and is less sensitive to the antilipolytic effect of insulin.8 This difference in adipocyte characteristics, combined with the proximity of the liver to intra-abdominal fat deposits, probably causes this organ to be more exposed to NEFAs than peripheral tissues.
Insulin sensitivity in turn regulates beta cell function, which is almost always decreased in obesity. NEFAs are important for normal beta cell function and enhance insulin release in response to glucose and other secretagogues. Chronic elevation of plasma glucose and NEFAs is harmful for beta cells, and is called glucolipotoxicity.7
To sum up, hyperinsulinism is common in obesity, and insulin resistance is characteristic wherever there is a significant weight increase.
Adipose tissueThe identification and characterization of leptin in 1994 strongly established adipose tissue as an endocrine organ.9 Adipose tissue is known to express and secrete a variety of bioactive peptides, called adipokines, acting at both local (autocrine/paracrine) and systemic (endocrine) levels. The significant endocrine function of adipose tissue is emphasized by the adverse effects of both its excess and deficiency. Excess adipose tissue or obesity, particularly in the visceral compartment, is associated with insulin resistance, hyperglycemia, dyslipidemia, arterial hypertension, and prothrombotic and proinflammatory states. The prevalence of obesity and these comorbid conditions, so-called metabolic syndrome, has reached epidemic proportions. Interestingly, adipose tissue deficiency, known as lipodystrophy, is also associated with characteristics of metabolic syndrome in both humans and rodents. Among proteins secreted by adipose tissue, leptin is the best characterized.10,11
Leptin (whose name derives from the Greek word leptos, which means thin) is a 16kDa polypeptide that contains 167 amino acids and shows structural homology to cytokines. The leptin gene was identified in 1994,9 and the leptin receptor in 1995.12 Adipocytes secrete leptin in direct proportion to adipose mass and nutritional status, and this secretion is greater from subcutaneous as compared to visceral fat mass. Leptin secretion per gram of adipose tissue is twice greater in obese as compared to slim people.
Although leptin was initially considered as an antiobesity hormone, its main role is to serve as a metabolic signal of energy sufficiency, rather than excess. Leptin levels rapidly decrease with calorie restriction and weight loss. This decrease is associated with physiological responses of adaptation to hunger, including increased appetite and decreased energy expenditure. These same responses are seen in leptin-deficient mice and humans despite massive obesity. Such responses are also corrected after the administration of low leptin doses. On the other hand, the common forms of obesity are characterized by high circulating leptin levels. The mechanisms of leptin resistance are unknown, but may result from defects in leptin signal or transport through the blood–brain barrier.10 In humans, mutations in leptin or its receptors are extremely rare.13
Obese people appear to be resistant or insensitive to leptin because despite the presence of high leptin levels, which should decrease food intake and body fat, they continue to have high body fat levels.
Other hormones secreted by adipose tissue having significant metabolic effects include adiponectin, resistin, TNFα, interleukine-6 (IL-6), proteins of the renin-angiotensin system, adipsin, acylation stimulating protein (ASP), and macrophage and monocyte chemoattractant protein-1 (MCP-1).
Adiponectin is secreted by adipose tissue only and is an abundant protein in plasma. Except in cases of severe malnutrition and in newborns, a strong negative correlation exists between plasma adiponectin levels in humans and fat mass.13 Adiponectin levels are decreased in obesity, but increase with weight loss. Adiponectin has been shown to improve insulin sensitivity in genetic or diet-induced obesity models.13
The peptide hormone, resistin (or FIZZ3), was initially identified as being produced by adipocytes only, and has been shown to play a significant role in insulin resistance induced by obesity.14 Resistin is expressed in rodent adipocytes14 and in human macrophages. Resistin levels increase with intake and in obesity, and decrease with PPARγ ligands.15
Retinol binding protein-4 (RBP4) is a 21kDa protein initially reported as an adipokine by Kahn et al.16 RBP4 has been found to induce insulin resistance by altering insulin signal in muscle16 and adipocytes.17
Pituitary glandIn obesity, the most obvious change in the hypothalamic–hypophyseal system is related to growth hormone (GH). Growth hormone secretion mainly depends on the interaction between GHRH and somatostatin. Ghrelin, the endogenous ligand of the GH secretagogue receptor, probably plays a role also.18 In addition, many neurotransmitters, peripheral hormones, and metabolic signals influence GH secretion.19
GH secretion is decreased in obesity. In both children and adults, the higher the body mass index, the lower the GH secretory response to different secretory stimuli,20 including the response to GHRH.21 It has been noted that GH secretion decreases by up to 6% per each unit increase in BMI at a given age. This relative GH deficiency in obesity may contribute to maintaining obesity. All defects in the GH-IGF1 in obesity are apparently reversible with weight loss, either induced by diet or as a result of surgery.22 When obese subjects are treated with high-dose GHRH and GHRP-6, a mean peak GH response of 40μg/L occurs, a massive response for obese subjects.20 The fact that somatotrophs, which have been at functional rest for years or decades of obesity, respond to those combined stimuli suggests a normal hypothalamic trophic hormone function and that somatotrophs do not become atrophic in obesity.20 The main cause of impaired GH secretion in obesity may be an impaired hypothalamus, an abnormal pituitary function, or a disturbance in the peripheral signals acting at the pituitary or hypothalamic level.
In obese people, GH secretion induced by exogenous administration of both GHRH or GHRP-6 is blocked, which rules out an endogenous GHRH secretory deficiency or the natural ligand of the GH secretagogue receptor as causative factors.
High insulin levels decrease GH release, and hyperinsulinemia commonly found in obesity may be related to this impaired GH secretion.22,23
Administration of a low dose of recombinant IGF-1 (rhIGF1) inhibits somatotroph response to GHRH in both obese and normal subjects, suggesting that somatotroph sensitivity to the inhibitory effect of rhIGF1 is preserved in obesity. This makes it less likely that GH hypersecretion in obesity is due to a greater than normal somatotroph inhibition induced by circulating IGF-1.
There is some evidence to suggest that decreased GH secretion in obesity is not related to the high leptin levels in obese people.24
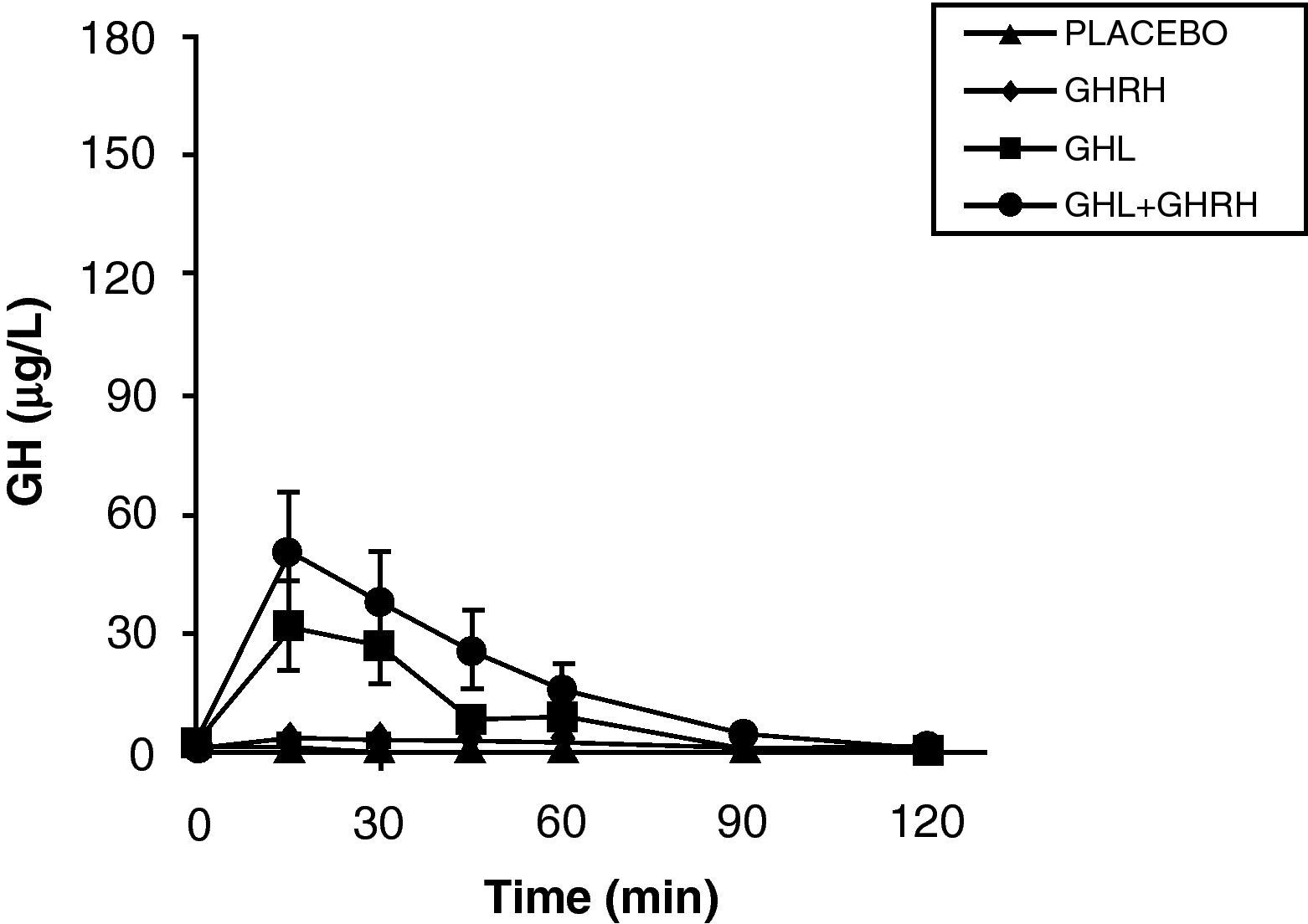
The reduction of free fatty acid (FFA) levels by acipimox, a drug that decreases lipids with minimal side effects, markedly increases GH secretion induced by pyridostigmine, GHRH, and GHRH+GHRP-6, restoring the level of this secretion to 50–70% of normal.25 These and other results suggest that high FFA levels play a significant role in GH decrease in obesity, and that treatment with lipolysis inhibitors may be helpful in restoring somatrotropic function.25 On the other hand, arginine and pyridostigmine, a drug that reduces somatostatinergic tone, increase GH secretion induced by GHRH in obese subjects.21 This, combined with an almost normal GH secretion in response to hypoglycemia in obese individuals, suggests that an increased somatostatinergic tone may partly explain the impaired somatotroph function in obesity. Although the mechanism responsible for GH hypersecretion in obesity is probably multifactorial,26 it has been noted that, in obese subjects, ghrelin is so far the most potent stimulus of GH secretion, and that massive GH secretion occurs following combined administration of ghrelin and GHRH (Fig. 1).
 mean±SE after administration of GHRH, ghrelin (GHL), or GHL+GHRH to obese patients.")
The persistence of a lower response as compared to normal subjects following the administration of ghrelin alone or combined with GHRH suggests that there is another defect involved in the impaired GH secretion found in obesity. Massive GH secretion after the administration of GHRH and ghrelin had not previously been found with any stimulus, and clearly suggests that impaired GH secretion in obesity is a functional and potentially reversible state, and that a decrease in ghrelin secretion could be at least partly responsible for GH hypersecretion in obesity. Plasma ghrelin levels are decreased in obese as compared to normal humans.27
Synthetic GH secretagogues (GHS) may be considered as ghrelin analogues,28 although all the actions of these agents known to date cannot be automatically transferred to ghrelin. Our results in obese subjects agree with the evidence that GHS induce a greater GH release than GHRH.29 We have previously studied GH secretion in obese people following stimulation with GHRP-6, a synthetic GHS. Although there were significant methodological differences between both studies, obese subjects showed a greater response to ghrelin than to the synthetic secretagogue GHRP-6, both alone and combined with GHRH. These results suggest that ghrelin is a more potent stimulus of GH release than synthetic GHS. As previously assumed, based on the results of synthetic GHS,20 additive effects of ghrelin and GHRH suggest that such peptides act, at least partly, by different mechanisms of action.
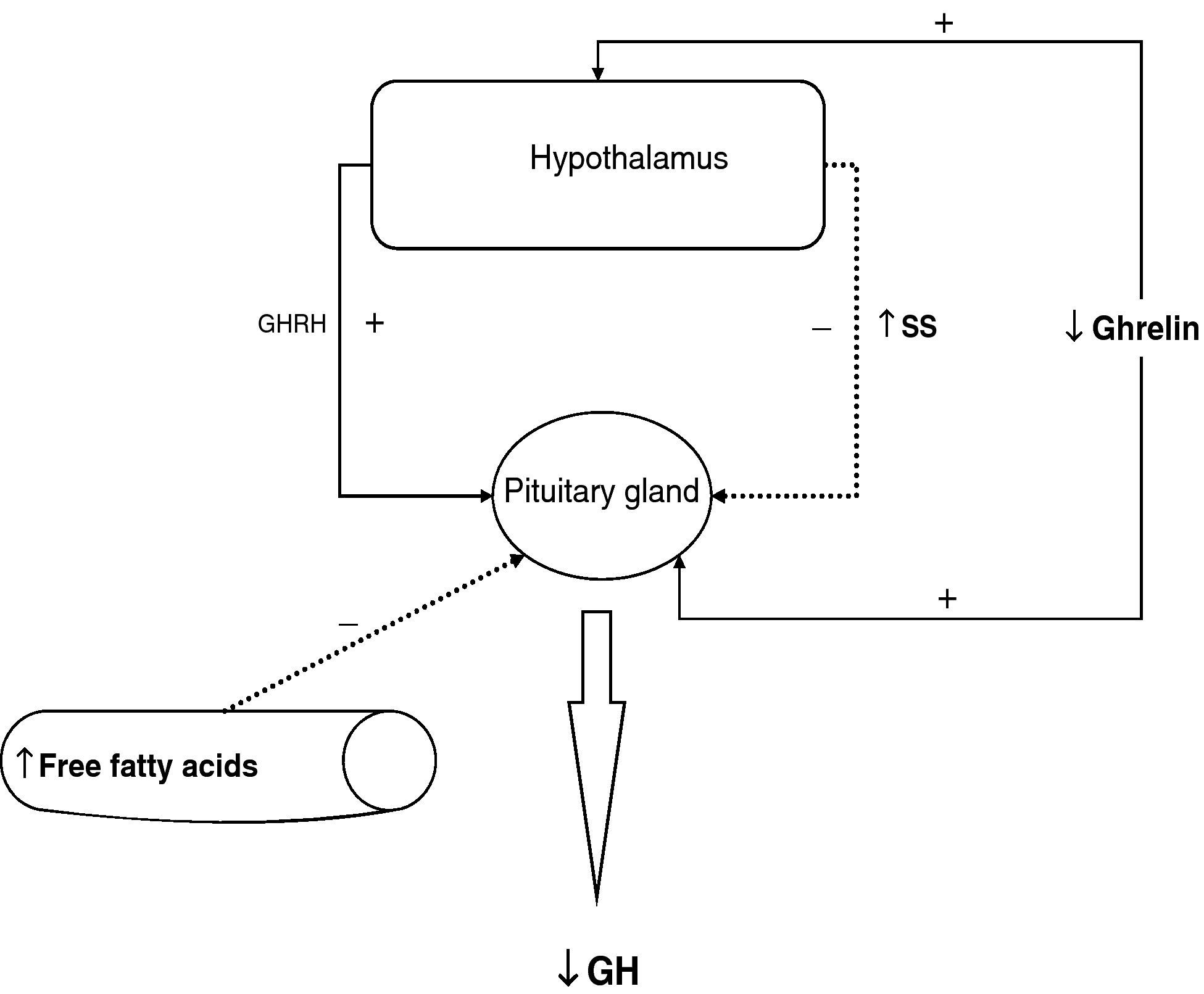
To sum up, growth hormone secretion is decreased in obesity, and impaired somatotropic function in obesity is functional and may be reversed in different situations. The pathophysiological mechanism responsible for this GH hypersecretion is probably multifactorial. Chronic somatostatin hypersecretion, elevation of plasma levels of free fatty acids and decreased ghrelin levels play an essential role, and some minor change in somatotropic cells may possibly coexist (Fig. 2).

Stimulation tests are required to diagnose GH deficiency in adults. Insulin-induced hypoglycemia (insulin tolerance test, ITT) is the test of choice for diagnosing GHD in adults. ITT is a potentially dangerous test. It is also contraindicated in many common clinical conditions in which GH deficiency may be suspected, such as ischemic heart disease, epilepsy, and advanced age.
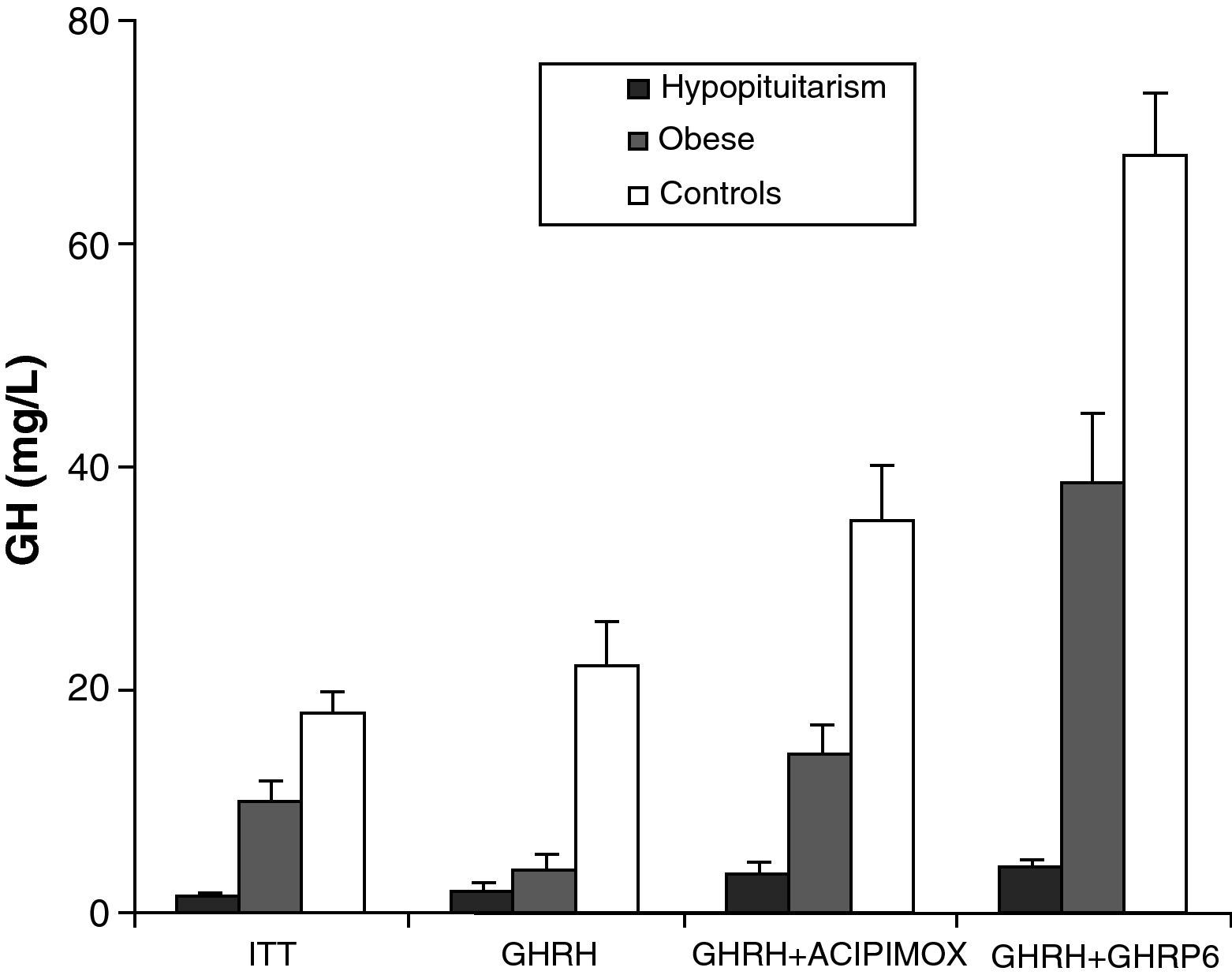
Obesity is probably the greatest confounding factor for diagnosis of GH deficiency in adults. Impaired GH secretion in obesity is known to parallel body composition changes such as increased visceral fat and decreased lean mass and bone mineral density. The response of normal obese subjects to different stimuli has been tested and compared to the response of obese patients with hypopituitarism to the same stimuli.30 Both groups showed similar responses to the administration of GHRH. However, GH response following the administration of GHRH and acipimox was markedly decreased in obese adults with hypopituitarism as compared to normal obese subjects. It may be concluded from these results that GH secretion following GHRH and acipimox is decreased in obese adults with hypopituitarism as compared to normal obese. The GHRH and acipimox test is safe and has no side effects. It could therefore be used to diagnose GH deficiency in adults.21 The maximum response to GHRH+GHRP-6 administration found in patients with hypopituitarism was lower than the minimum response seen in normal and obese subjects. After ITT, GHRH, or GHRH+acipimox, however, the maximum response in patients with hypopituitarism was lower than the minimum response in normal subjects, but greater than the minimum response in obese subjects. We also found a greater differential between normal or obese subjects and patients with hypopituitarism for GHRH+GHRP-6 as compared to ITT, GHRH, or GHRH+acipimox30 (Fig. 3). We showed with these studies that the administration of both acipimox or GHRP-6 reverses the functional hyposomatotropism of obesity after GHRH, but that these agents are unable to reverse the organic hyposomatotropism of hypopituitarism. Combined administration of GHRH and GHRP-6 is the test that best differentiates both conditions without the side effects of insulin-induced hypoglycemia.30
GonadsOvary (mean±SE) after insulin-induced hypoglycemia (ITT), GHRH, GHRH+acipimox, and GHRH+GHRP6 in patients with hypopituitarism (, HIPO), obese patients, (¿) and normal controls (□).")
In women, obesity occurs as a number of conditions preceding metabolic abnormalities such as prediabetes, diabetes, and cardiovascular disease.31 Such conditions include early menarche, infertility, and polycystic ovary syndrome (PCOS). On the other hand, at the end of the reproductive life of women, ovarian function suppression is also associated with the development of obesity, because menopause precipitates abdominal weight gain and is associated with multiple adverse metabolic consequences.
In women, obesity is associated with significant changes in plasma steroid levels. The secretion rates of testosterone, dihydrotestosterone, and androstenedione are higher in women with morbid obesity. Body distribution of fat mass is another significant factor. Sex hormone production and metabolism are different in morbid obese women with different phenotypes. Women with abdominal obesity have an increased androgen production rate and higher free testosterone and estradiol levels, while women with a gluteofemoral fat distribution produce a high amount of estrone due to peripheral estrogen aromatization. Other studies have reported that in premenopausal obese women, an increase in visceral fat is significantly associated with decreases in sex hormone binding globulin (SHBG) levels and the 17-beta estradiol/free testosterone ratio and to elevated free testosterone levels after adjusting for age and total fat mass. In women, visceral fat loss is associated with increases in SHBG levels and the 17-beta estradiol/free testosterone ratio regardless of the total amount of fat lost.32 PCOS is the most common ovarian pathology in premenopausal women, and obesity is a frequent occurrence in this endocrine disease. Depending on the population studied, 20–69% of women with PCOS are obese (BMI>30), and regardless of obesity, women with PCOS have an increased accumulation of intra-abdominal fat.
After menopause, the dramatic drop in estrogen levels, combined with relative hyperandrogenism, contributes to weight gain and to changes in adipose tissue distribution.
TestisMale obesity is associated with reductions in total testosterone and SHBG levels.33,34 In the Massachusetts Male Ageing Study, men who were obese at baseline and during follow-up, as measured by BMI or central obesity (waist circumference or waist–hip index), had a greater decrease in total and free testosterone and SHBG as compared to men who had never been rated as obese.35 The pathogenetic factors related to these decreased testosterone levels in obesity include decreased SHBG binding capacity, reduced LH pulse amplitude, and hyperestrogenemia.
Assessment of the sex hormone profile of obese subjects shows that plasma testosterone levels are moderately or greatly decreased in obese men despite a reduction in SHBG levels.36 In obese men, androgen decrease has been found to represent a continuum that may be seen with any degree of obesity. An inversely proportional relationship exists between plasma total testosterone, free testosterone, and SHBG, and visceral fat mass. Isidori et al.37 examined the relationship between leptin concentrations and sex hormone levels in adult men and found that circulating leptin levels correlated to total and free testosterone even after adjusting for SHBG, LH, and estradiol, and that leptin was the best hormonal predictor of decreased androgen levels in obese men.
The prevalence of metabolic syndrome increases with age and is independently associated with lower androgen levels and higher estrogen levels.38 In multivariate models, age, waist circumference, and health state were associated with low androgen levels and symptomatic androgen deficiency, and waist circumference was the most important associated factor of all variables.39
Adrenal glandBecause of the relationship between obesity, central fat distribution, and high cortisol levels (Cushing's syndrome), many studies have attempted to determine whether cortisol plays a role in the development of obesity in the general population, central fat distribution in men and women, and changes in body composition with age. Urinary free cortisol levels (per gram of creatinine) are significantly increased in obese women with abdominal fat distribution as compared to those with peripheral fat distribution or normal controls. Basal cortisol and ACTH levels significantly increase following the administration of CRF in all groups, but this increase is significantly greater in women with abdominal obesity as compared to those with peripheral fat distribution or normal controls. These results therefore suggest that obese women with abdominal fat distribution have hyperactivity of the hypothalamic–hypophyseal–adrenal axis (HHA).40
Although plasma cortisol levels are generally assumed to be normal in metabolic syndrome and obesity, there are exaggerated dynamic cortisol responses.41 Abdominal obesity is particularly associated with increased urinary free cortisol excretion and increased total cortisol production rates.42 These data suggest that small changes occur in obesity in the feedback mechanism, in inhibitory feedback of the HHA, and that there is an impaired peripheral clearance at liver level, with minor differences between the manifestations of metabolic syndrome. This, in turn, suggests that excess cortisol production may contribute to the development of obesity. However, there is no adequate evidence to conclusively state that increased cortisol secretion is the cause of excess adipose tissue deposition in humans. Cortisol secretion in obesity has been quantified by different methods, including isotopic dilution and measurements of free urinary cortisol and C21 metabolites. These studies have invariably shown an increased cortisol production in obesity.43 Recent studies have found similar increases in cortisol production in obese women with polycystic ovary syndrome and normal control obese women.44
Local conversion of cortisone into cortisol by 11-beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) has been found to be significantly related to local activation of the glucocorticoid receptor, which provides an explanation for the parallelism between idiopathic and cushingoid obesity. In humans, 11β-HSD1 mRNA activity is increased in abdominal subcutaneous adipose tissue from obese subjects both in vivo and in vitro in most studies.45 There are studies suggesting that 11β-HSD1 mRNA levels are sometimes also increased in visceral omental adipose tissue from obese women and are a strong predictor of adipose tissue size in this visceral fat deposit.46 An increase in cortisol production in the liver, not associated with systemic hypercorticoidism because it is compensated for by an increased hepatic catabolism of cortisol and a lower activity of the adenocortical axis, has been reported in obese patients with metabolic syndrome.47
Gastrointestinal hormonesGhrelin is a 28-amino acid peptide hormone mainly produced in the stomach and whose structure has a fatty acid chain in the third amino acid of the N-terminal end which is essential for some of its biological actions, including its potent stimulating activity on somatotropic cells. Ghrelin stimulates growth hormone secretion by acting directly at the pituitary level through the GH secretagogue receptor.20,29 In addition to stimulating GH secretion, ghrelin has other endocrine and nonendocrine actions: it stimulates lactotropic and corticotropic secretion, inhibits the gonadal axis, stimulates appetite and positive energy balance, controls gastric motility and acid secretion, acts in both an endocrine and exocrine pancreatic function, has cardiovascular actions, influences behavior and sleep, and modulates cell and immune system proliferation. X/A cells in oxyntic glands of the stomach are the main source of circulating ghrelin. Ghrelin is also synthesized in the small bowel, but to a lesser extent, and the amount of ghrelin produced decreases as the distance from the pylorus increases.
The active form of ghrelin appears to need the fatty acid in the third amino acid of the N-terminal end for its endocrine actions. The non-acylated form has no endocrine activity,28 but is not biologically inactive because it shares with the acylated form antiproliferative effects on prostate cancer cell lines, has a negative inotropic effect on cardiac papillary muscle, and stimulates adipogenesis in bone marrow, although the transduction mechanisms of these effects need to be studied. Ghrelin appears to have a role in the metabolic and neuroendocrine response to food intake. Its circulating levels are increased in anorexia and cachexia and decreased in obesity,48,49 and plasma ghrelin levels are inversely related to body mass index, body fat mass, plasma leptin levels, and glucose and insulin levels.50
There are various studies which suggest that ghrelin may play a significant role in feeding and weight control. The GH secretagogue receptor (GHS-R1a) in the arcuate nucleus is involved in the regulation of GH secretion, food intake, and adiposity.51 Circulating ghrelin levels increase with fasting and decrease after nutrient intake in both mice and humans.52,53 Cummings et al.53 showed preprandial peaks and postprandial suppression of ghrelin levels in humans at the start of voluntary food intake. These data suggest that ghrelin plays a role at the start of intake.53 Various types of evidence suggest a role for ghrelin in the long-term regulation of body weight and energy homeostasis. In humans, circulating ghrelin levels are inversely related to the degree of adiposity. Lower levels are found in obese subjects, and elevated levels in conditions such as anorexia nervosa, malignancies, or cachexia associated with chronic heart failure.48,49 Ghrelin levels increase after weight loss achieved both with diet alone or with a combination of diet and exercise, and are suppressed with overnutrition and during treatment of anorexia nervosa.54 Circulating ghrelin levels under fasting conditions and after food intake are impaired in hepatic insufficiency and restored after liver transplantation.55,56 The neutralization of circulating ghrelin levels impaired weight recovery in an animal model.57
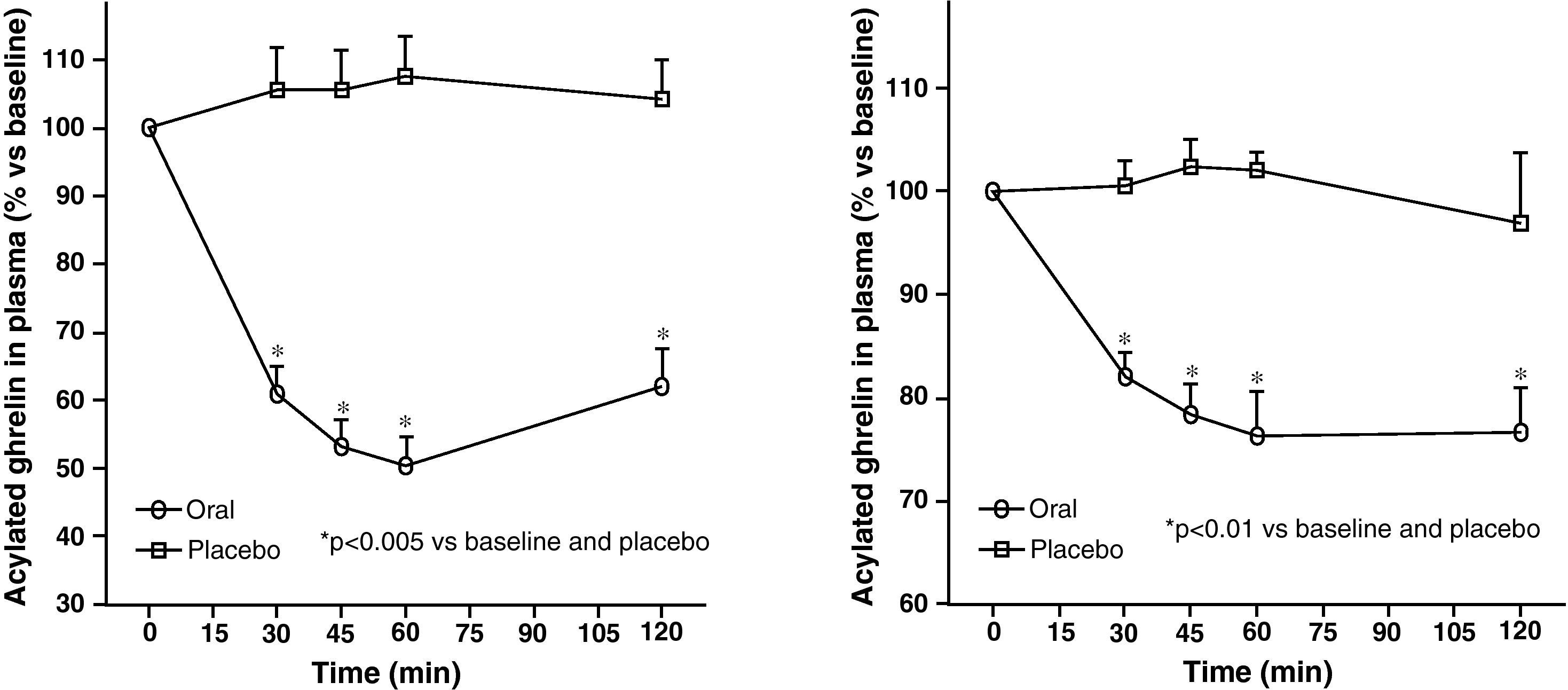
Circulating plasma ghrelin levels increase before a meal and decrease after nutrient intake (Fig. 4). Gastric distention occurring with water infusion into the stomach does not result in a reduction of ghrelin levels. However, intake of non-nutritive fiber does decrease ghrelin levels. It has been shown that obese subjects do not experience the decrease in plasma ghrelin levels seen in slim people following food intake.58 Although obese people with Prader-Willi syndrome, characterized by hyperphagia and obesity, have elevated ghrelin levels, most obese subjects show lower fasting ghrelin concentrations as compared to normal weight volunteers59–61. Insulin resistance has been postulated as being involved in these lower ghrelin levels in the obese. The increase in plasma ghrelin levels with diet-induced weight loss is consistent with the hypothesis that ghrelin plays a role in the long-term regulation of body weight in humans. Gastric bypass is associated with markedly suppressed ghrelin levels, which probably contribute to the weight-reducing effect of this procedure.62 To sum up, ghrelin, the only circulating orexigenic hormone, is decreased in obesity.

Peptide YY (PYY) is a 36-amino acid peptide. PYY is synthesized and released into the circulation by specialized enteroendocrine cells called L cells, which are mainly located in the distal gastrointestinal tract. Two main forms of PYY have been reported, PYY1-36 and PYY3-36. Circulating PYY levels increase in response to nutrient intake, with caloric load, food consistency, and nutrient composition affecting its circulating levels. An initial postprandial increase in circulating PYY levels is seen 15min after food intake, before the nutrients reach L cells in the distal gastrointestinal tract. Hormonal and neuronal mechanisms are therefore implicated in this initial release. PYY levels typically peak 1–2h after intake, and this peak is followed by a phase of several hours with stable levels.63 In 2002, Batterham et al. reported that peripheral administration of PYY3-36 decreased food intake in rodents and humans of normal weight.64 Subsequent studies confirmed that this anorectic effect of PYY3-36 was preserved in obese subjects. In addition to contributing to postprandial satiety, several current lines of evidence suggest that PYY3-36 plays a role in the long-term regulation of body weight.65 PYY3-36 is selective for the Y2 receptor (Y2R). The crucial role of this receptor as a mediator in the anorectic effect of PYY3-36 during feeding was initially identified by Batterham et al., who found that mice lacking the Y2 receptor were resistant to the anorexigenic effects of PYY3-3664.
PP is a 36-amino acid peptide mainly produced by F cells, located in the periphery of the islets of Langerhans in the pancreas and to a lesser extent in the colon. Circulating PP concentrations increase after nutrient intake in a biphasic manner and in proportion to the caloric load, and remain elevated for approximately 6h following intake. There is evidence to suggest that PP plays a role in body weight regulation in rodents, but the role of PP in the pathogenesis of non-syndromic obesity is not clear.64
GLP1 (glucagon-like peptide type 1), produced in L cells in the small bowel and secreted in response to nutrients, is known as an incretin hormone. GLP1 is released into circulation approximately 30min after nutrient intake. Its main effect is to stimulate glucose-dependent insulin release by the pancreatic islets. GLP1 restores first and second-phase insulin secretion in response to glucose.66 In addition, GLP1 inhibits glucagon release after intake, slows gastric emptying, and decreases food intake. Treatment with GLP-1 and its analogues is associated with weight loss partly because of their effects upon gastric emptying and their gastrointestinal side effects of nausea and vomiting.
Oxyntomodulin (OXM) is a 37-amino acid peptide hormone initially isolated from porcine jejunoileal cells. OXM is released by L cells in response to food intake and in proportion to caloric load. Chronic central administration of OXM for a 7-day period decreases food intake, weight gain, and adiposity in rats with no evidence of tachyphylaxis. In humans with normal body weight, OXM reduces immediate calorie intake by 19%. This inhibitory effect lasts for approximately 12h after intake.
Amylin, a 37-amino acid peptide, is a neuroendocrine hormone. It is cosecreted with insulin by pancreatic beta cells in response to nutrient intake, incretin hormones, and neural impulse. Amylin has glucoregulatory actions that complement insulin actions, suppresses postprandial glucagon secretion, and delays gastric emptying.
Thyroid glandHypothyroidism is normally associated with a modest weight gain and decreased thermogenesis and metabolism, while hyperthyroidism is related to weight loss despite increased appetite and metabolism.
Although obese subjects usually have a normal thyroid function, TSH and BMI are known to be positively correlated. In fact, many studies of children, adolescents, and adults have shown slightly increased TSH levels in obese individuals as compared to normal slim subjects.67 Increased TSH levels were not related to iodine deficiency or autoimmune thyroiditis in various studies.67 By contrast, obesity has been found to increase susceptibility to autoimmune thyroid disease, with an emerging role of leptin as a peripheral determinant.68 A neuroendocrine dysfunction leading to abnormal TSH secretion has been suggested as the reason for elevated TSH levels in obesity. In particular, leptin has been shown to alter the hypothalamic–hypophyseal axis.69 The TRH test, however, ruled out an impaired pituitary response in obese children with increased TSH levels.70 On the other hand, increased TSH levels suggest hormone resistance. Increases in TSH and peripheral thyroid hormones may be an adaptation process to increased basal energy metabolism, and thus to energy expenditure. In agreement with this theory, elevated TSH levels in obesity are normalized after any substantial weight loss.67 A moderate increase in TSH levels in obesity is associated with normal T4, normal free T4, and moderately increased free and total T3, as well as an increased thyroid volume.67 Free T3 levels are increased in obesity due to changes in the monodeiodination pathways. An elevation in thyroid hormone concentrations in obesity possibly increases energy expenditure and prevents energy accumulation as fat. Since fasting and weight loss are associated with decreased thyroid hormone levels and thus with a drop in basal energy expenditure, this fact probably contributes to the difficulties in maintaining any weight loss. The physiological pathways linking obesity to the increase in thyroid hormones are not clear. Leptin may be one of the links between body weight and thyroid hormones.67 Additional studies are needed to determine whether a mild thyroid hormone deficiency and a resultant increase in TSH, for example to the upper normal limit, are related to obesity development.71 On the other hand, although thyroid hormones have frequently and improperly been used to achieve weight loss in euthyroid obese subjects, they are not indicated for body weight control, except for hypothyroid obese subjects.71
Conflicts of interestThe authors state that they have no conflicts of interest.
Studies mentioned in this article have been funded by: Grants PI070413 and PI10/00088 from the FIS of Instituto de Salud Carlos III, and grants PS07/12, INCITE08ENA916110ES, INCITE09E1R91634ES, IN845B-2010/187 from Xunta de Galicia.
We thank Ramón Pensado for his technical assistance.
Please, cite this article as: Álvarez-Castro P, et al. Función endocrina en la obesidad. Endocrinol Nutr. 2011; 58:422–32.
