


Familial hypocalciuric hypercalcemia (FHH) is a condition of autosomal dominant inheritance due to inactivating mutations in the calcium-sensing receptor (CaSR) gene. It is biochemically characterized by mild to moderate hypercalcemia, relative hypocalciuria, and inappropriately normal or high (by 20%) PTH levels. Clinically, patients usually have few or no symptoms of hypercalcemia, and bone or renal involvement is rare. Adequate differential diagnosis between primary hyperparathyroidism (PHP) and FHH is important, because the latter is a benign condition that does not improve after parathyroidectomy. We report here on the members of a family with FHH showing the biochemical heterogeneity of the disease and the difficulty of differential diagnosis with PHP.
Measurements of intact PTH (normal range, 10–65pg/mL) were performed by electrochemoluminescence with an ELECSYS analyzer. A Cobas 711 analyzer (Roche Diagnostics) was used to perform measurements of phosphorus (normal range, 2.7–4.5mg/dL) by spectrophotometry using molybdate; calcium (normal range, 8.6–10.4mg/dL) using the Schwarzenbach method with o-cresolphthalein complexone; and creatinine (normal range, 0.67–1.17mg/dL) by the Jaffé method. Urine measurements of calcium (normal range, 100–320mg/24h), phosphorus (normal range, 700–1500mg/24h), and creatinine (normal ranges, 740–1570mg/24h in females and 1040–2350mg/24h in males) were performed using the same methods as in serum. 25-hydroxyvitamin D levels were normal in all reported cases (normal range, 30–100ng/mL). Calcium levels were expressed corrected for albumin [4-albumin (g/dL)×0.8]+calcium (mg/dL). Magnesium, gastrin, growth hormone, insulin-like growth factor-1, TSH, free T4, ACTH, cortisol, prolactin, LH, FSH, subunit alpha, and calcitonin were measured in all subjects, as well as catecholamines in 24-h urine, with results within the normal range in all cases. Orthopantomography ruled out the presence of fibro-osseous jaw tumors, and renal ultrasound showed kidneys of normal morphology in all the subjects studied. The genetic study showed no mutations associated with multiple endocrine neoplasia type 1 syndromes in any of the cases.
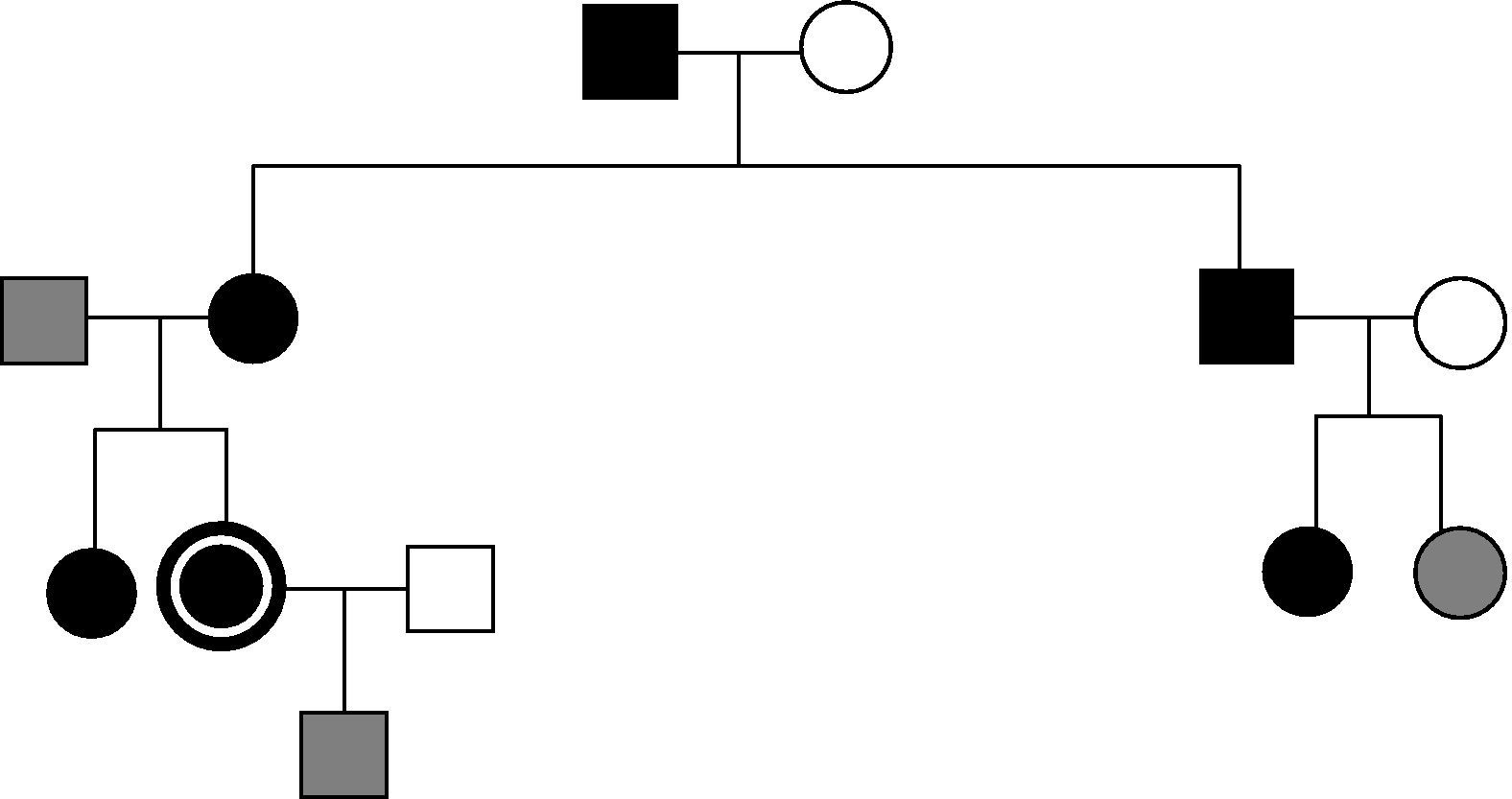
The index case was a 16-year-old female patient referred in 1996 for hypercalcemia work-up. From that date, nine members of the family have been studied, of whom six are affected. Among the nine family members, six were tested at the hospital. The four family members affected and followed up at our center are reported below (Fig. 1). The remaining three subjects were seen at another center (their data are not available).

The index case reported urinary frequency, polydipsia, and epigastric pain. Laboratory test results were as follows in 1996 and in the last measurement respectively: calcium, 10.6 and 11.1mg/dL; phosphate, 2.4 and 3.6mg/dL; and PTH, 40.7 and 62.2pg/mL. The corresponding results in 24-h urine included: calcium, 127.9 and 179.4mg/24h; phosphorus, 680 and 870mg/24h; and Ca/CrCl<0.01 in all measurements. A 99m-Tc sestamibi scan showed no pathological uptake. Bone densitometry (BMD) performed at 23 years was normal for age.
The 20-year-old patient's sister was symptom-free and had the following blood test results: calcium, 10.5 and 10.9mg/dL; phosphorus, 2.9 and 3.4mg/dL; and PTH, 48.6 and 94.7pg/mL. The results in 24-h urine included: calcium, 174.4 and 231.4mg/24h; phosphorus, 710 and 1320mg/24h; and Ca/CrCl<0.01 in all measurements. BMD measured at 26 years showed osteopenia in lumbar spine, femur, and forearm.
The 70-year-old maternal grandfather was symptom-free and had the following blood test results: calcium 11.4mg/dL, phosphorus 2.4mg/dL, and PTH 69.9pg/mL. His 24-hour urine results included calcium 348.2mg/24h, phosphorus 1240mg/24h, and Ca/CrCl>0.01.
The blood test results of the 40-year-old mother, asymptomatic and premenopausal, included: calcium, 10.8 and 11.6mg/dL; phosphorus, 3 and 3.1mg/dL; and PTH, 83.4 and 118.3pg/mL. The results in 24-h urine included: calcium, 228 and 496.8mg/24h and phosphorus, 710 and 1004mg/24h, Ca/CrCl>0.01 in all measurements, except for one in which it was lower than 0.01. BMD showed osteopenia in the femur and forearm and osteoporosis in the lumbar spine. Sestamibi revealed hyperplasia in all four parathyroid glands. A molecular study of the CaSR gene was performed in the mother using automatic sequencing of exons 2 (codons 1–62), 3 (codons 62–164), 4 (region 5′: codons 165–250), and 7 (region 5′: codons 578–887), and no mutation was found in the areas studied. Based on the presence of data consistent with PHP in the mother and grandmother (measurements in the latter could only be performed once and follow-up could not be continued), they were diagnosed with PHP and parathyroidectomy was indicated for the mother. The upper left (540mg) and right (580mg, normal 150–300mg) glands were resected, while the other glands could not be seen. Histology showed mild hyperplasia. Since then, the patient has had high calcium levels in plasma and urine. Three years after surgery, a genetic study extension revealed a previously unreported mutation in exon 7 c.3163_3170delinsTTACTGGGGA (p.Val1055fs). The two daughters were subsequently studied and were found to have the same mutation as the mother in heterozygosis, but this was not present in the grandson.
FHH results from inactivating mutations (causing function loss) in CaSR, whose gene is encoded for in the long arm of chromosome 3. The presence of such mutations of parathyroid glands and renal tubules is the basis for FHH.1 FHH should be considered in individuals with asymptomatic persistent hypercalcemia. The characteristic feature for differentiating FHH from PHP is calcium excretion in 24-h urine, which is typically less than 200mg in patients with FHH. By contrast, 40% of patients with PHP have hypercalciuria. Fractional calcium excretion has been reported to be the most helpful parameter for discriminating between the two conditions. Approximately 80% of patients with FHH have Ca/Cr<0.01,2 while a similar or higher proportion of patients with PHP have higher values3; however, the Ca/Cr ratio may be <0.01 in approximately 20% of patients with PHP, particularly when this is associated with vitamin D deficiency. Differentiating between FHH and PHP on the basis of biochemical measurements is not always possible, and it has recently been reported that some members of families with FHH may have hypercalciuria and values of Ca/CrCl>0.02.4,5 Hypercalcemia in other family members suggests FHH, but isolated familial PHP may also exist. When biochemical and clinical data do not allow for differential diagnosis, a genetic study of the CaSR gene is indicated. More than 200 mutations have been reported to date in this gene (http://www.casrdb.mcgill.ca). However, as shown by this case, the genetic study is not infallible either. It has been reported in recent years that no mutations in the CaSR gene are found in up to one third of families with FHH, and the possibility of mutations in sequences regulating gene function has been suggested. In the reported family, the presence of hypercalciuria in its two older members, the bone impact shown, and the initial negative genetic study of the mother led to the erroneous diagnosis of familial PHP and to the resultant indication of parathyroidectomy in one of them. The long clinical follow-up of this family for 15 years finally allowed for a diagnosis of FHH. In conclusion, the differentiation of FHH of atypical presentation from PHP may be difficult, but it is essential in order to prevent ineffective surgical procedures.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Mijares Zamuner MB, et al. Hipercalcemia hipocalciúrica familiar: una presentación atípica. Endocrinol Nutr. 2013;60:270–1.
