


McCune-Albright syndrome (MAS) is a heterogeneous, uncommon condition caused by postzygotic, somatic, and sporadic mutation of the GNAS gene, encoding the stimulatory alpha subunit (αs) of the G protein-coupled receptor.1 Clinically, this syndrome consists of a triad characterized by bone fibrous dysplasia, café-au-lait spots and hyperfunctioning endocrinopathies such as early puberty, hyperthyroidism, excess growth hormone (GH), hyperprolactinemia, and hyperadrenocorticism. However, a diagnosis of MAS is made when two of the three clinical signs are present.1,2
We report the case of a 16-year-old male patient from Mérida (Venezuela) with no family history and a personal history of multiple femoral fractures secondary to polyostotic fibrous dysplasia diagnosed at three years of age who attended the endocrinology unit for tall height. He also reported continuous sever holocranial headache and hyposmia which had intensified in recent years.
Physical examination revealed 93kg of weight (greater than the 97° percentile), 183cm of height (greater than the 97° percentile) with a genetic height potential (sum of the height of both parents+12.5cm/2) of 169±10cm, a body weight index (IMC) of 26.6kg/m2, and 120/70mmHg of blood pressure. Café-au-lait spots 5.5 and 7cm in diameter, both with irregular margins, were seen in the back of the neck and the right buttock respectively. He also had craniofacial deformity characterized by macrocephalia, frontal prominence and hypertelorism, and external supports in both lower limbs due to multiple fractures. An android distribution of body hair was also noted, with testes of 25ml.
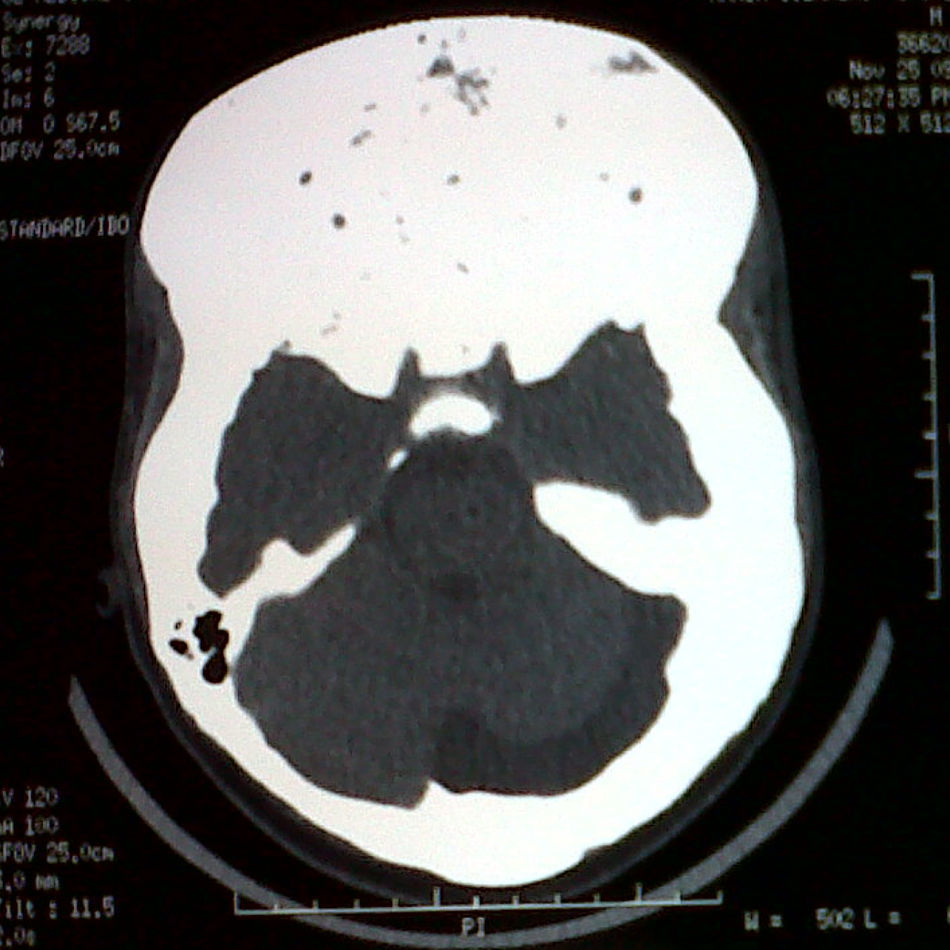
Clinical laboratory tests showed normal blood glucose, as well as the following results: calcium 8.8mg/dL (normal range (NR), 8.7–10.3), magnesium 2.1mg/dL (NR, 1.40–2.40), phosphorus 2.2mg/dL (NR, 2.7–4.5mg/dL), parathyroid hormone 35.1pg/mL (NR, 10–69), alkaline phosphatase 1396.1IU/L (NR, 98–271), basal somatotropin (GH) 7.1μg/L (NR, 0–2.5), GH of 5.4μg/L two hours after a 75g glucose load (NR, less than 1), IGF-1 725ng/mL (NR, 72–385), normal thyrotropin and free thyroxine, basal cortisol 8.7μg/dL (NR, 5–25), prolactin 57ng/mL (NR in males, 0–15), follicle-stimulating hormone 0.35mIU/mL (NR, 0.7–11), luteinizing hormone 0.2mIU/mL (NR, 0.8–7.6), and total testosterone 92.8ng/dL (NR, 286–1511). X-rays of the left hand and wrist revealed a bone age of 15 years. A computed tomography scan showed increased bone mass volume in the cranial vault and structures of the frontal, orbital, and mastoid regions consistent with fibrous dysplasia, and a thickening of sellar region bones, which prevented adequate visualization of the pituitary gland (Fig. 1). Pituitary magnetic resonance imaging with gadolinium could not be performed due to external supports and intramedullary nails in the lower limbs. Computed campimetry was therefore performed, showing reliability indices and a normal bilateral foveal threshold. However, slightly decreased retinal sensitivity, enlarged blind spot, and superior temporal focal scotoma consistent with prechiasmatic lesion were seen in the visual field of the right eye. The left eye in turn had slightly decreased retinal sensitivity, with a blind spot of normal size and no scotomas.

Based on the clinical and laboratory findings and imaging tests, octreotide LAR 20mg once monthly, cabergoline 0.5mg twice weekly, testosterone undecanoate 1000mg every three months, and zoledronic acid 5mg once a year were prescribed. At a new visit at three months, the patient reported a slight improvement in headache and showed a basal GH value of 2.4μg/L. Other test results included: GH of 2.5μg/L two hours after a 75g glucose load, 2.5μbasal IGF-1 105ng/mL, prolactin 37ng/mL, total testosterone 1290ng/dL, and alkaline phosphatase 1126.2IU/L. Based on these results, the monthly dose of octreotide LAR was increased to 30mg, with no change in any of the other medical indications.
This patient had MAD due to the coexistence of polyostotic fibrous dysplasia, café-au-lait spots, and endocrinopathies such as excess GH and hyperprolactinemia. The prevalence of this disease is unknown, but it is estimated to occur in 1/100,000 to 1/1,000,000 newborns, being more common in females.1 Fibrous dysplasia may be monostotic, affecting a single bone, or polyostotic when more than two bones are involved. The first form is more common and occurs in 7% of benign tumor bones.1,2
Approximately 20% of patients with MAS have excess GH. A high prevalence of concomitant hyperprolactinemia (71–92%) is found in these patients. Thirty-three percent of these cases are due to a pituitary microadenoma, and all the others to hyperplasia of mammosomatotroph cells.3 Excess GH is particularly harmful in patients with MAS because it may accelerate fibrous dysplasia, especially in craniofacial bones, potentially causing vision and hearing loss.3 First line treatment of excess GH and hyperprolactinemia is medical, consisting of the use of somatostatin analogs and dopamine D2 receptor agonists, because if pituitary adenoma occurs, (transsphenoidal) surgery is not effective due to massive thickening of the skull base resulting from craniofacial fibrous dysplasia3; it should be noted, however, that the response of this group of patients to medical treatment with cabergoline and octreotide LAR is usually consistent but inadequate, and hormone control criteria are not reached in most cases.4 According to the consensus on acromegaly cure criteria published in 2010, disease control is defined as random GH levels less than 1μg/L with normal IGF-1 levels for sex and age. In the event of disagreement between these tests, it is recommended that the GH level be measured two hours after a 75g glucose load. This should be less than 0.4μg/L.5 It should be noted that when this patient was seen again three months after the start of treatment with octreotide LAR, he was found to have a basal GH of 2.4μg/L, with a GH level two hours after the load of 2.5μg/L, which despite the normal IGF-1 level revealed an inadequate response to the somatostatin analog and led to the dose being increased to 30mg monthly.
The most common endocrinopathy in MAS is early puberty, but some cases of central hypogonadism have exceptionally been reported in both sexes.6,7 In the reported patient, the cause must have been acquired because he showed good virilization and adequate testicular volume. Of note in the functional history was hyposmia, which is commonly associated with hypogonadism in Kallmann syndrome, but may possibly be due in this case to severe cranial sclerosis obliterating the cribriform cartilage of the ethmoid bone, thus damaging the olphactory fibers. While this patient had high prolactin levels, these were not so high as to explain hypogonadism. It is therefore likely that the cause could be related to gonadotroph cell damage due to fibrous dysplasia of the sella turcica.
Fibrous dysplasia results from GNAS gene mutation and causes abnormal proliferation and differentiation of osteoblasts together with increased osteoclastic activity. This mutation is also associated with increased expression of fibroblast growth factor 23, which increases renal phosphate excretion, causing hypophosphatemia and aggravating the bone mineralization defect.2,8 It should be noted that this patient had mild hypophosphatemia upon admission. Based on pathophysiological understanding of the disease, bisphosphonates have been increasingly used in recent years. Pamidronate and zoledronic acid are the most commonly used bisphosphonates.9,10 The prognosis of polyostotic disease is usually good, but tends to be worse in patients with MAS having excess GH.2 Monitoring is based on bone turnover markers such as alkaline phosphatase, which are elevated in active disease and tend to decrease in response to antiresorptive treatment.2
Please, cite this article as: Lima-Martínez MM, et al. Hipogonadismo hipogonadotropo en un varón con síndrome de McCune-Albright. Endocrinol Nutr. 2013;60:145–7.
