


Adverse events during intrauterine life may program organ growth and favor disease later in life. This is the usually called ‘Barker's hypothesis’. Increasing evidence suggests that conditions such as vascular disease, hypertension, metabolic syndrome, and type 2 diabetes mellitus are programmed during the early stages of fetal development and become manifest in late stages of life, when there is an added impact of lifestyle and other conventional acquired environmental risk factors that interact with genetic factors. The aim of this review was to provide additional, updated evidence to support the association between intrauterine fetal health and increased prevalence of chronic non-communicable diseases in adulthood. Various potential cellular and molecular mechanisms proposed to be related to the above hypothesis are discussed, including endothelial function, oxidative stress, insulin resistance, and mitochondrial function.
Cambios metabólicos in utero establecen patrones fisiológicos y estructurales a largo plazo que pueden «programar» la salud durante la vida adulta, teoría popularmente conocida como «Hipótesis de Barker». Evidencia experimental y clínica sugiere que patologías como hipertensión arterial, enfermedad isquémica coronaria, síndrome metabólico y diabetes mellitus tipo 2, pueden «programarse» durante las primeras etapas del desarrollo fetal y manifestarse en etapas tardías, al interactuar con el estilo de vida y otros factores de riesgo adquiridos convencionales con el medio ambiente. El objetivo de esta revisión es presentar evidencia adicional y actualizada que apoyen la asociación entre la salud fetal intrauterina y el aumento en la prevalencia de enfermedades crónicas no transmisibles en la edad adulta. La función endotelial, el estrés oxidativo, la resistencia a la insulina, y la función mitocondrial son tratadas como posibles mecanismos celulares y moleculares.
Over the past decades, it has been suggested in various research areas that events involved in normal fetal development have long-term effects and influence health during adult life.1–7 It is thought that in utero metabolic changes may establish long-term physiological and structural patterns that “program” health during adult life.2–4 The studies of Barker et al.5–7 in the 80s established that the prevalence of some adult diseases, such as atherosclerosis, high blood pressure (HBP), stroke, type 2 diabetes mellitus, and dyslipidemia was related to intrauterine environment (“Barker's hypothesis”). This hypothesis is currently known as the ‘Developmental Origins of Health and Disease hypothesis’ (DOHaD).6,7
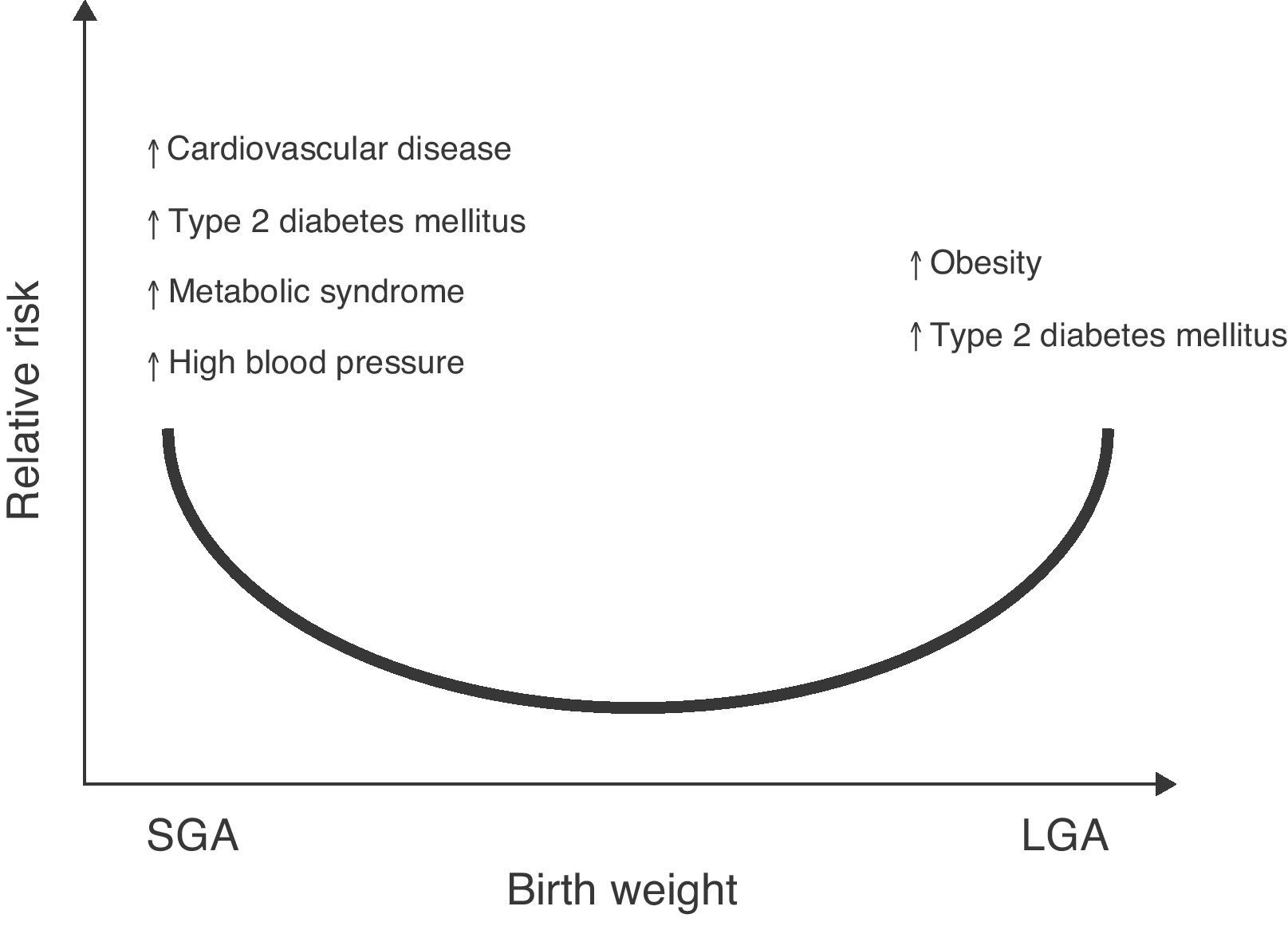
The association of low birth weight and height with an increased risk of a subsequent occurrence of diseases such as high blood pressure (HBP), metabolic syndrome, and stroke has also been reported.7,8 Changes in body weight or composition at birth, either in the upper normal range for gestational age (large for gestational age, LGA) or significant reductions in birth height and weight (small for gestational age, SGA) may lead to metabolic sequelae in adult life9,10 (Fig. 1).

Relative risk of non-communicable chronic diseases in adult life depending on birth weight. Based on epidemiological and experimental observations and on “Barker's hypothesis”,5–7 there is adequate evidence to confirm that an increase in weight for gestational age (LGA: large for gestational age) increases the risk of obesity and metabolic syndrome in the postnatal stage.121 Similarly, a low weight for gestational age (SGA: small for gestational age) would suggest an increased risk of type 2 diabetes mellitus, cardiovascular disease, metabolic syndrome, high blood pressure, and obesity in adult life.122
Additional research has subsequently supported this relationship, although some of these mechanisms have still to be elucidated.11,12 Experimentally, nutritional restriction during pregnancy has been shown to irreversibly affect the structure, metabolism, and function of some organs, thus “programming” the offspring for future diseases.13,14
Mechanisms related to fetal programmingThe hypothesis concerning origins related to fetal programming states that coronary disease, stroke, HBP, and type 2 diabetes mellitus have their origin in the plasticity of development which occurs in response to maternal and placental factors during fetal life and breast-feeding. For example, it has been postulated that HBP may be due to the lower number of glomeruli in people with small size such as infants with low birth weight (LBW) and intrauterine growth retardation (IUGR).15,16
The second process involves hormonal and metabolic regulation. A premature LBW newborn is more prone to have a “thrifty” metabolic pattern (the thrifty phenotype hypothesis) for nutrient management.5–7 Insulin resistance and an increased oxidative stress (OS) state, which is associated with LBW, could be considered from this perspective as a persistent fetal response for glucose preservation in the brain at the expense of the transport of this carbohydrate to muscle for metabolism and growth. According to the “thrifty” phenotype hypothesis, a poor fetal growth would decrease the number of pancreatic β cells and insulin-producing capacity, leading to insulin resistance states in adult life.17 There is consistent evidence that newborns with LBW and IUGR will experience insulin resistance.5–7 A systematic review published in 2008 by Whincup et al.18 reported that in most populations studied, birth weight was inversely related to the risk of suffering type 2 diabetes mellitus and HBP.
A third association between LBW and fetal programming is that people with low height at birth are more vulnerable to adverse environmental influences later in life.19,20 However, no clear association exists between high weight and an increased risk of HBP.21,22
Nutrition during pregnancy, birth weight, and metabolic programmingAnimal experiments and epidemiological observations in humans suggest that nutrition received in the intrauterine environment modulates the function of several tissues with metabolic activity in postnatal life.23,24 A significant consequence of calorie and nutrient restriction is the apparently accelerated postnatal growth period.25 In humans, the relationship between poverty and malnutrition during pregnancy increases childhood morbidity and mortality.7 In this regard, Barker et al.26 assessed glucose tolerance in subjects aged 64 years. Their results showed a strong association between low birth weight and metabolic diseases. Newborns weighing less than 2500g at birth had a 7.4-fold greater risk of suffering type 2 diabetes mellitus or glucose intolerance as compared to those weighing more than 4000g.
It has also been seen that catch-up growth occurs in approximately 85% of premature or LBW children in the first three years of life. Singhal et al.27,28 and Ross et al.29 found an association between early fast postnatal growth and an increased risk of having metabolic syndrome indicators and an unfavorable body composition. A similar hypothesis is the catch-up growth hypothesis proposed by Arends et al.30 and Cianfarani et al.31. The basis of this hypothesis is that newborns with nutritional restriction have lower levels of insulin and insulin-like growth factors such as IGF-1 and IGF binding protein 3 (IGFBP-3). While normalization of the levels of these hormones usually occurs during the first three months of postnatal life, coinciding with the rapid growth seen in these newborns, tissues with previous chronic insulin and IGF-1 deficiency suddenly facing high concentrations of these hormones may possibly develop insulin resistance as a defense mechanism against hypoglycemia. This would explain the associations found between catch-up growth and an increased risk of metabolic syndrome and postnatal obesity.29–33
In addition to an impaired body composition resulting from maternal nutritional restriction, increased intramuscular triglyceride (IMTG) levels have been found in infants with impaired intrauterine growth. This may partly explain their predisposition to type 2 diabetes mellitus.32 Changes in IMTG levels are associated with a decreased activity of the enzyme carnitine palmitoyl transferase-1 (CPT-1b), a key protein in the oxidation of these fatty acids.33
In addition, adequate placentation and fetal development also depend on levels of other important hormones such as placental leptin34 and adiponectin.35 In postnatal life, these hormones are mainly secreted by adipose tissue and are involved in the regulation of metabolism, cardiovascular function, and energy homeostasis, among others. In adults, leptin regulates long-term energy consumption and expenditure, while adiponectin has anti-inflammatory properties and increases insulin sensitivity.36 Increased leptin levels have been found in fetuses and placentas from diabetic mothers, while decreased adiponectin levels have been seen in their children at birth. In newborns with IUGR, the levels of these two adipokines are decreased.37 However, it has been noted that at one year of age these children have increased leptin levels as compared to children with adequate birth weight, and adults with a history of LBW have been found to have increased leptin levels as compared to subjects with similar body weight indices.38 In rats subjected to malnutrition in utero, leptin increase in the early postnatal stages has been associated with obesity and metabolic syndrome indicators in adult age.39 Moreover, extremely low and high birth weights have been related to higher fat percentages later in life.40 Adipokine levels in early developmental stages possibly play a significant role in programming the body composition of individuals. In humans, hyperleptinemia states have been seen in obesity, metabolic syndrome, and cardiovascular disease in subjects with LBW.41
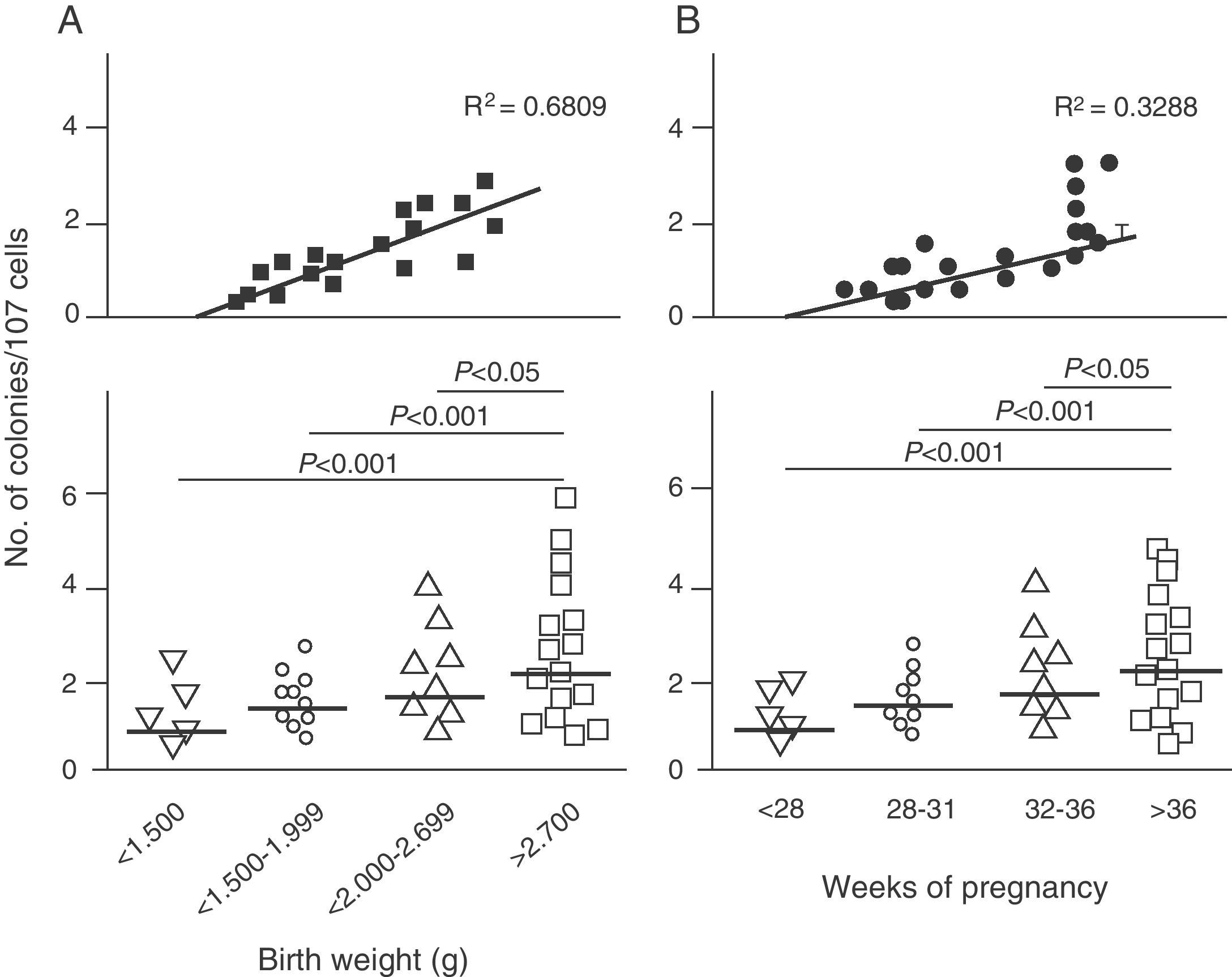
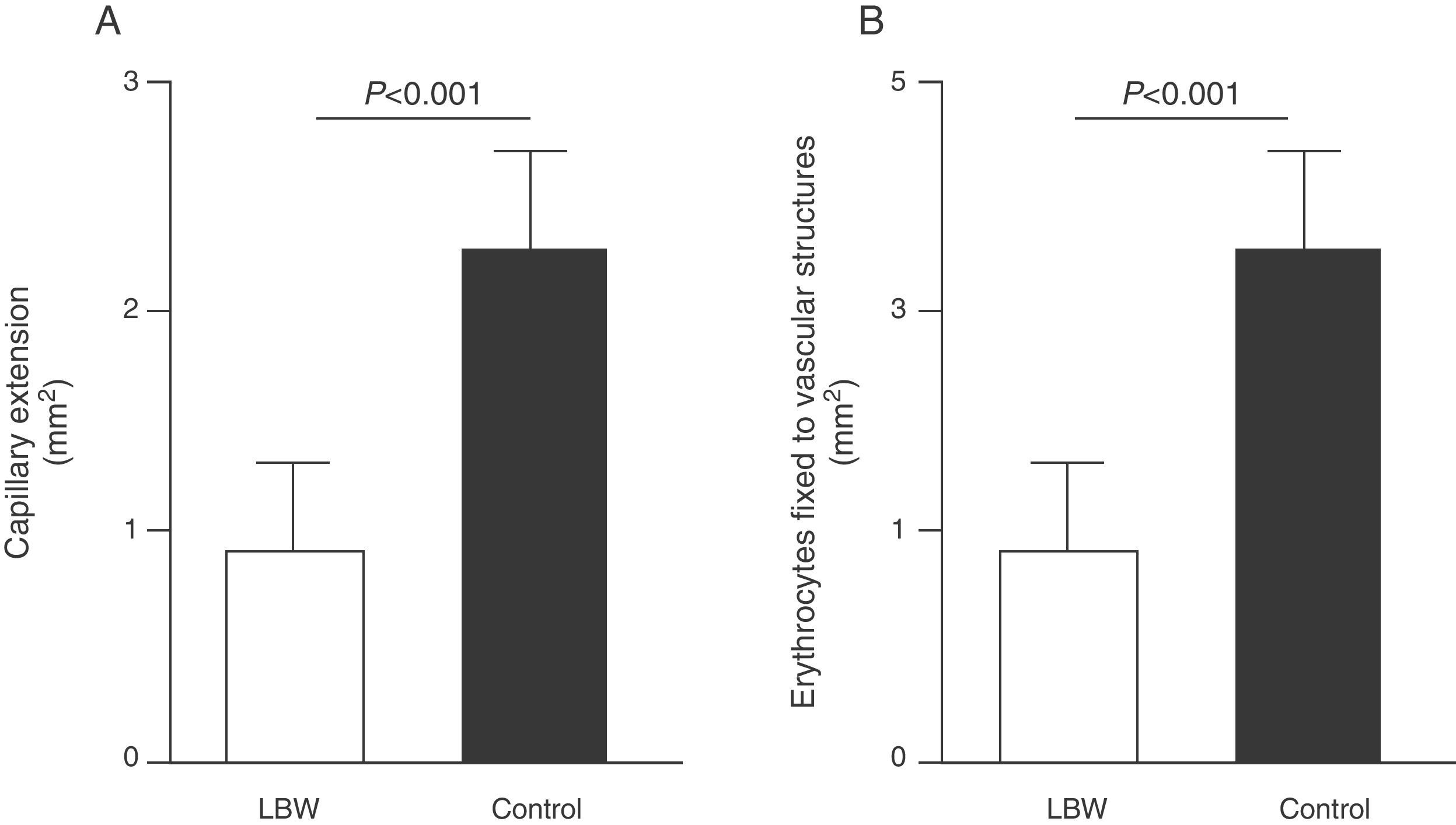
Endothelial dysfunction and adult hypertensionMultiple mechanisms mediate the fetal programming of adult HBP.42–44 For example, several authors have shown relationships with changes in kidney function (a decreased number of nephrons), the neuroendocrine system (dysregulation of the hypothalamic–pituitary–adrenal axis), and the vascular system (vascular dysfunction and decreased arteriole and capillary density).45,46 Ligi et al.47 recently focused their attention on the origin of vascular abnormalities and the endothelial angiogenic properties of endothelial colony-forming cells (ECFCs) present in umbilical cord blood from LBW infants as compared to term infants born after normal delivery. ECFCs are cells characterized by their ability to form endothelial cell colonies in vitro and to repair the damage caused by a change in vascular phenotype (Fig. 2A and B). Thus, Ligi et al.47 reported that cultured colonies of ECFCs from LBW infants had a decreased ability to form tubular and capillary structures, less proliferation capacity, and a lower angiogenic potential (Fig. 3A and B).
 on endothelial angiogenic properties of endothelial colony-forming cells (ECFCs) present in umbilical cord blood. As shown, cultured ECFCs from infants with LBW had a lower capacity to form colonies (A). This finding was also made in relation to gestational time and birth weight (B). Taken and modified from Ligi I, et al. Blood. 2011;118:1699–709.")
Effect of low birth weight (LBW) on endothelial angiogenic properties of endothelial colony-forming cells (ECFCs) present in umbilical cord blood. As shown, cultured ECFCs from infants with LBW had a lower capacity to form colonies (A). This finding was also made in relation to gestational time and birth weight (B). Taken and modified from Ligi I, et al. Blood. 2011;118:1699–709.
 on endothelial proliferation properties of endothelial colony-forming cells (ECFCs) present in umbilical cord blood. As shown, cultured ECFC colonies from infants with LBW showed a reduced capacity to form tubular and capillary structures (A), less proliferation capacity, and less angiogenic potential (B). Taken and modified from Ligi I, et al. Blood. 2011;118:1699–709.")
Effect of low birth weight (LBW) on endothelial proliferation properties of endothelial colony-forming cells (ECFCs) present in umbilical cord blood. As shown, cultured ECFC colonies from infants with LBW showed a reduced capacity to form tubular and capillary structures (A), less proliferation capacity, and less angiogenic potential (B). Taken and modified from Ligi I, et al. Blood. 2011;118:1699–709.
The above findings provide the first evidence in humans of a relationship between birth weight and the angiogenic properties of ECFCs, and a potential mechanism of microvascular aberration, arteriolar narrowing, and decreased angiogenesis previously reported in animal models.48–50 Interestingly, these researchers found a strong inverse correlation between birth weight and “angiogenic defects” in ECFCs in children with birth weights lower than 1500g (Fig. 2A). This finding is consistent with several epidemiological studies showing a correlation between the risk of HBP in young adults and the degree of immaturity at birth.51,52
Nitric oxide and fetal programmingSodium retention in rats with prenatally programmed hypertension may also result from an imbalance in nitric oxide (NO) bioavailability. In kidney, NO plays many important roles such as the regulation of renal hemodynamics, the maintenance of medullary perfusion, the modulation of tubuloglomerular response, and tubular sodium reabsorption, resulting in a net effect of natriuresis and diuresis.53
Cavanal et al.54 measured NO production in aortic segments from infants born to diabetic mothers. Basal NO production was found to be significantly depressed in the group of infants born to diabetic mothers as compared to controls. After stimulation with acetylcholine (ACh) or bradykinin (BK), NO production significantly increased in all groups, but a greater increase was seen in controls. On the other hand, decreased angiotensin 1–7 (ANG 1–7) levels in the kidney may also interfere with NO production and bioavailability, as suggested by Li et al.55. These authors reported that the vasodilating effect of ANG 1–7 on aortic rings from mice was completely abolished by pre-treatment with l-NAME, a NO synthase inhibitor, suggesting that endothelial NO is involved in the vasodilating effect of ANG 1–7 in this experimental model.
The exact mechanism for the occurrence of HBP in children born to mothers with metabolic changes during pregnancy is not fully understood. However, several mechanisms may contribute to the development of HBP in adults, which reinforces the need for close monitoring of maternal and placental metabolism during pregnancy to prevent permanent changes in homeostasis in the offspring.
Low birth weight and its relation to vascular function and oxidative stressIt should be noted that both LBW and premature birth induce changes in vascular development due to the immaturity of several biological systems which are modulated by the intrauterine and extrauterine environments. The significance of this environmental change, as a key event in the etiopathogenesis of vascular dysfunction, lies in the inappropriate growth of blood vessels normally developing in infants born at term. Among many other differences, normal extrauterine environment is markedly hypoxic as compared to the extrauterine environment. During pregnancy, the fetus gradually prepares for transition to the relatively oxygen-rich extrauterine environment, as is shown by the substantial increase in antioxidant enzyme levels during the last weeks of pregnancy.56 If preterm delivery occurs (particularly before 32 weeks), this preparation is not completed, and the fetus is susceptible to environmental factors such as elevated oxidative stress (OS).57
In addition to OS status, excess exposure to fetal glucocorticoids may increase the risk of developing hypertensive disorders, thus conferring a greater risk of cardiovascular comorbidity in adult life.58 Children with LBW have been shown to have in adult life an independent risk of experiencing psychiatric and cardiovascular diseases associated with the dysregulation of placental levels of 11β-HSD (11β-hydroxysteroid dehydrogenase).59 The decreased regulation of 11β-HSD levels increases fetal exposure to maternal glucocorticoids60 and, as an adaptive response, accelerated intrauterine fetal maturation occurs, as it has recently been shown by Roghair et al.61. These authors also explored the early origins of HBP in a rat model treated with a 11β-HSD inhibitor (CBX: carbenoxolone) during the last week of pregnancy. Blood pressure increase was shown to be associated with vascular dysfunction and exaggerated superoxide anion (O2−) production in male adults, but not in females, in pups exposed to CBX. Although excess glucocorticoids are known to induce vascular O2− production62 and OS has been associated with programmed vascular dysfunction,63,64 these interactions have not been evaluated from a developmental perspective.
There is however a pending question: how does this environmental change exactly affect vascular characteristics in children born preterm or with LBW? A possibility is that the angiostatic capacity seen in newborns with LBW is the result of early exposure to the extrauterine environment, and is therefore not yet operative. If this is true, one may speculate from the molecular viewpoint that LBW induces defects in gene expression profiles resulting in decreased angiogenesis and angiostatic imbalance. Such changes would include: (a) the dysregulation of molecules with angiostatic properties such as e-NOS and AKT,65 (b) the downregulation of matrix metalloproteases (MMP-2 and MMP-9)66 that degrade basal membrane and allow for the migration of ECFCs, and (c) the inappropriate regulation of vascular growth factors (VEGF, FGF, PGF), an increase in molecules associated with tumor proliferation capacity (PLXDC-1),67 and a decrease in cytokines with angiogenic activity (CXCL-1 and CXCL-5)68 (Fig. 4).
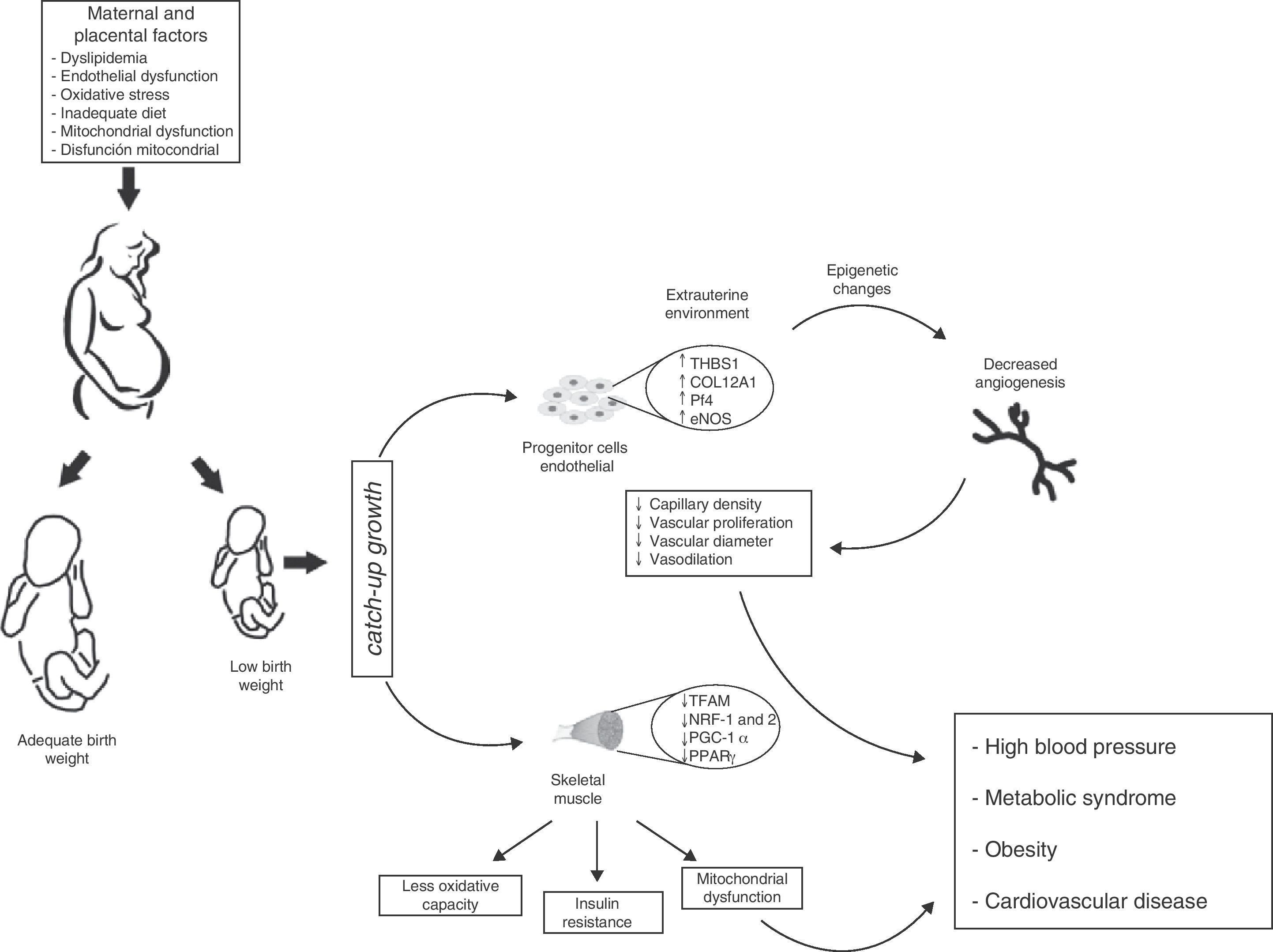
 have less angiogenic potential (↓ capillary density, ↓ vascular diameter, ↓ endothelial cell proliferation, and ↓ vasodilation). These adaptive vascular changes are reflected in transcriptional changes in metabolic pathways and vascular growth. Some of these changes result from modification of epigenetic gene regulation (TSBS1, COL12A1, Pf4, eNOS). In skeletal muscle, changes in the intrauterine environment (fetal hypoxia) induce epigenetic changes in genes with postnatal metabolic function (TFAM, NRF-1 and 2, PGC-1α, PPARγ) leading to decreased oxidative capacity, mitochondrial dysfunction, and insulin resistance. Infants with LBW therefore have higher blood pressure and an increased risk of HBP, obesity, metabolic syndrome, and cardiovascular disease in adulthood.")
Proposed model of intrauterine fetal health programming and its consequences for health in adult life. Endothelial stem cells from infants with low birth weight (LBW) have less angiogenic potential (↓ capillary density, ↓ vascular diameter, ↓ endothelial cell proliferation, and ↓ vasodilation). These adaptive vascular changes are reflected in transcriptional changes in metabolic pathways and vascular growth. Some of these changes result from modification of epigenetic gene regulation (TSBS1, COL12A1, Pf4, eNOS). In skeletal muscle, changes in the intrauterine environment (fetal hypoxia) induce epigenetic changes in genes with postnatal metabolic function (TFAM, NRF-1 and 2, PGC-1α, PPARγ) leading to decreased oxidative capacity, mitochondrial dysfunction, and insulin resistance. Infants with LBW therefore have higher blood pressure and an increased risk of HBP, obesity, metabolic syndrome, and cardiovascular disease in adulthood.
Therefore, an altered gene expression pattern is consistent with vasculogenetic defects, a lack of anastomoses, less vasodilation, greater smooth muscle proliferation, and endothelial dysfunction. However, these findings do not fully prove a causative and direct relationship of HBP to vascular defects in postnatal life, but they do suggest its contribution to an impaired angiogenic potential of ECFCs, as shown by Ligi et al.47 (Fig. 4).
Insulin resistance and vascular and metabolic programming in adultsThe conditions altering vascular function during fetal and neonatal life, such as hyperglycemia,69 gestational diabetes,70,71 insulin resistance,72 or hyperoxia73 promote the development of cardiovascular, metabolic, and endocrine diseases in adult life.74–76 However, their relationship with an inadequate prenatal environment due to maternal metabolic changes is still controversial. Amri et al.77 showed a decreased number of nephrons in the offspring of rats with gestational diabetes treated with streptozotocin (STZ) at the start of pregnancy. In other studies, Rocha et al.78 and Magaton et al.79 examined the effect of diabetes mellitus induced in rats before mating on the blood pressure and kidney function of their offspring. Although the results showed increased blood pressure levels, no changes in the number of nephrons were reported after glomerular isolation. By contrast, Tran et al.80 showed in diabetic Tg-Hoxb7 mice that kidneys from newborn pups from diabetic dams had glomeruli with a smaller functional area and a relative reduction in the number of nephrons. This result agrees with the one reported by Ortiz et al.,81 who studied rats treated with dexamethasone at different periods during pregnancy and found a decreased number of glomeruli in treated pups.
In addition to HBP, an increased risk of obesity82 and type 2 diabetes mellitus83 have been shown in children from mothers with gestational diabetes. Silverman et al.84 assessed the offspring of women with pregestational diabetes mellitus (type 1 and type 2) and gestational diabetes. The prevalence of glucose intolerance and insulin resistance was shown to be significantly greater in these groups as compared to controls. Similar results were found by Pettitt et al.,85 who tested the effect of abnormal glucose tolerance on the offspring of Pima indigenous women during pregnancy. The authors correlated metabolic abnormalities occurring in diabetic pregnancy with insulin resistance states, HBP, obesity, and diabetes in the offspring. The mechanism by which maternal hyperglycemia increases metabolic risk in the offspring is yet to be fully elucidated. An increased glucose supply to the fetus may possibly act as a stimulus to improve insulin production and expose the fetus to hyperinsulinemia. It has also been postulated that increased fetal leptin production may contribute to the metabolic disorder in the postnatal stage.86,87 In critical developmental periods, increased levels of hormones such as insulin and leptin may cause an “endogenous dysfunctional teratogenic” metabolism.88 For example, pups from hyperglycemic rats develop “metabolic programming” of hypothalamic (neuropeptidergic) neurons, which increases their neuropeptide and orexigenic activity, possibly leading to a state of hyperphagia with a resultant weight increase.89
In addition to its effect on carbohydrate metabolism, insulin is involved in other functions, including the modification of lipid and protein metabolism, amino acid and electrolyte transport, cell cycle regulation, apoptosis, and NO synthesis.90,91 In addition to insulin, other hormones such as angiotensin II (ANG II) and norepinephrine may influence several biological processes related to HBP occurrence.92,93 ANG II affects insulin signaling through the SOCS-3 protein pathway. It has also been postulated that ANG II acts through the AT-1 receptor to decrease insulin-dependent NO production by the inhibition of ERK-1/2 proteins and the activation of JNK.94 On the other hand, ANG II increases NADPH oxidase activity through the AT-1 receptor, enhancing the generation of reactive oxygen species (ROS).95 Zhou et al.96 recently showed that the regulation of ANG II and oxidative stress are associated with endothelial dysfunction and insulin resistance in hypertensive patients sensitive to renal sodium excretion.
Renal sodium excretion is another significant factor which contributes to HBP occurrence in adults. Rocco et al.97 studied sodium excretion in diabetic offspring with and without sodium overload and found that, under normal conditions, children born to diabetic mothers had sodium excretion levels similar to the control group. However, after sodium overload, children from diabetic mothers did not achieve the sodium excretion levels of the control group. Nehiri et al.98 similarly examined sodium excretion in children of diabetic mothers who received a high sodium diet for three consecutive days. In the offspring of diabetic mothers, study of the renal cortex revealed an increased expression of the epithelial sodium channel (ENAC) and sodium/potassium ATPase (Na+/K+ ATPase), with no changes in sodium/hydrogen exchanger (NHE3) or other transporters, which suggests that sodium retention was due to an increased reabsorption from distal nephron segments.
In utero muscle development and insulin resistanceSkeletal muscle fibers show a heterogeneous molecular arrangement and are called type I, IIa, IIb, and IIx fibers depending on the type of metabolism. These types in turn express different physiological functions provided by the specificity of their myosin chain isoforms.99 At the beginning of fetal development, type I muscle fibers are mainly expressed, while type IIa, IIb, and IIx fibers develop in later gestational stages. In animal models with the same embryonic development as humans (e.g. sheep and swine), muscle fibers are mainly formed in the second half of pregnancy, while the expression of metabolic function is established before birth.100,101 Thus, it may be hypothesized that skeletal muscle is a key target for prenatal programming, because a change in the intrauterine environment may affect skeletal muscle composition and function in adulthood. In fact, several authors have shown that decreases in type I and IIa fibers in subjects with LBW are associated with a marked reduction in the oxidative capacity of muscle, a state which has been related to insulin resistance and obesity in adult life102,103 (Fig. 4). In parallel, Zhu et al.104 noted that the offspring of mothers given a diet restricted in nutrients and calories during the last trimester of pregnancy showed a decreased number of muscle fibers and a relative increase in the number of type IIb fibers as compared to the group fed ad libitum.
Thorn et al.105 recently studied a model of chronic placental insufficiency and showed an 80% increase in insulin receptor levels in skeletal muscle, while the expression of catalytic subunit of phosphatidylinositol 3-kinase (PI3 K) and protein kinase-B (Akt/PKB) was suppressed by 36% and 37% respectively. These changes suggest a metabolic adaptation strategy for survival in cases of IUGR, but with potentially harmful consequences in carbohydrate regulation and insulin sensitivity in postnatal life. In fact, in infants with IUGR, muscle levels of AKT-2, PI3-K, and GLUT-4 were shown to be lower as compared to those of children with normal growth.106 These findings suggest that although fetuses with IUGR have a greater number of insulin receptors in skeletal muscle, the low number of molecules responsible for the activation of GLUT-4 could partly explain the occurrence of insulin resistance and metabolic syndrome in adulthood.
Mitochondrial bioenergetics: the role of mitochondrionThe loss of mitochondrial membrane potential (Δψm) and the induction of transient mitochondrial permeability are closely related to mitochondrial events during apoptosis as events unique to cells,107 and to some complications which occur during pregnancy such as IUGR, prematurity, and LBW.108 The main consequence of the long-term opening of the transient mitochondrial permeability transition pore is depolarization due to the disappearance of the proton gradient, and the resultant inhibition of mitochondrial respiration. Under conditions of fetal hypoxia, excess ROS results in increased cytosolic Ca2+ levels, which accumulate in the mitochondrion, with the resultant loss of Δψm. ROS have been reported to be able to decrease mitochondrial biogenesis (MB) and impair mitochondrial function, causing placental mitochondrial dysfunction with a resultant impairment in tissue hypoxia and changes in placental development.109,110 This was seen by a decreased expression of various MB regulators, including mitochondrial transcription factor A (TFAM) and nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2). Bioenergetic and functional mitochondrial changes have also been seen in placentas from mothers of children with LBW.111,112 Ali et al.113 reported that, in endothelial cells, activation of PGC-1α induced the expression of hemoxygenase, a stress–response protein. Recently, Barrès et al.114 found a negative correlation between (non-CpG) cytosine hypermethylation of the PGC-1α promoter and protein expression and mitochondrial density in muscle cells from subjects with type 2 diabetes mellitus. In addition, mice which recovered their nutritional status showed a restoration of eNOS levels and MB in adipose tissue and muscle, as well as weight reduction as compared to normal mice.115
Similarly, MB defects and other defects related to its morphogenesis affect enzyme activity and decrease oxidative capacity, leading to increased intramyocellular lipid levels. When combined with the inability of mitochondrion to use these metabolic substrates, the accumulation of some metabolites harmful for its functioning (e.g. ceramides, diacylglycerol, and ROS) in skeletal muscle induces an impairment in insulin signaling pathways and a resultant reduction in glucose uptake.115 In addition, because of changes in the signaling pathways of some transcription factors inducing MB, such as PGC-1α and PPARγ, these have been found to be decreased as the result of changes in the intrauterine environment116,117 (Fig. 4). Therefore, impaired fatty acid uptake and oxidation, together with a lower antioxidant capacity and an increased OS state in postnatal life, characterize metabolic dysregulation in subjects with altered intrauterine growth. However, other mechanisms could be modulated by changes in the embryonic cell phenotype resulting from this metabolic adaptation.
Future prospectsEpidemiological observations by Barker et al.5–7 provided early evidence that adults with a prior coronary event or metabolic syndrome were small at birth and remained thin for their first three years of life. This delayed fetal growth, combined with a subsequent, disproportionate postnatal growth, was associated with insulin resistance and endothelial dysfunction in adult life. Skeletal muscle is the main site of carbohydrate and fatty acid metabolism, and any aberration in in utero muscle development will therefore facilitate disease expression in adulthood. Changes in muscle fiber distribution, changes in insulin signaling, a decreased angiogenic capacity, and an impaired mitochondrial function are also significant events in the development of cardiometabolic diseases in adults with LBW (Fig. 4).
The next step in the development of the potential fetal programming hypotheses described in this review will be experimental verification in humans in altered metabolic states such as preeclampsia, gestational diabetes, and pregnancy-induced hypertension, which are risk factors associated with the fetal programming of adult health. It would also be interesting to explore some signaling pathways related to mitochondrial biogenesis (PCG-1α, TFAM, NRF-1, and NRF-2), redox state (NFKβ), and changes in energy demand (MAPK). Very little is currently known about changes in vascular function and mitochondrial biogenesis in metabolic states related to the fetal programming of non-communicable chronic diseases. It has been postulated that such effects could play a role in the attenuation related to mitochondrial age and in the “dysfunction” of diseases related to oxidative metabolism such as myopathies, cancer, and mental disease.
Prolonged breast-feeding, delay in the introduction of supplemental feeding,118 protein intake in the first year of life, the practice of physical exercise during pregnancy,119 etc. are actual steps being taken nowadays in pediatrics, endocrinology, gynecology, and obstetrics.120
FundingThis study had no external funding source.
Conflicts of interestThe authors state that they have no conflicts of interest.
Please cite this article as: Ramírez-Vélez R. Programación Fetal in utero y su impacto en la salud del adulto. Endocrinol Nutr. 2012;59:383–93.
