


Mujer de 20 años que acude a urgencias por presentar desde hacia 3 días un cuadro de ansiedad, palpitaciones, ortopnea y disnea paroxística nocturna. No presenta edemas en miembros inferiores, ni disminución de la diuresis. No refiere cefalea, ni hipersudación, ni cuadro infeccioso en los días previos. Como antecedentes destaca, enfermedad de Von Hippel-Lindau (VHL) IIb, con amaurosis bilateral secundaria a angiomas retinianos múltiples desde los 14 años. No se realizó el cribado y seguimiento del síndrome ya que por motivos personales la paciente decidió no acudir a las revisiones periódicas.
La exploración clínica destaca estado general regular. TA 125/70mmHg; FC 120 lpm; SatO2 96% (FiO2 21%); Temperatura 36,5°. En la auscultación cardiaca se objetivan ruidos rítmicos, con soplo sistólico panfocal grado IV/VI, más audible en foco mitral e irradiado hacia axila. Resto de la exploración clínica por aparatos y sistemas dentro de la normalidad.
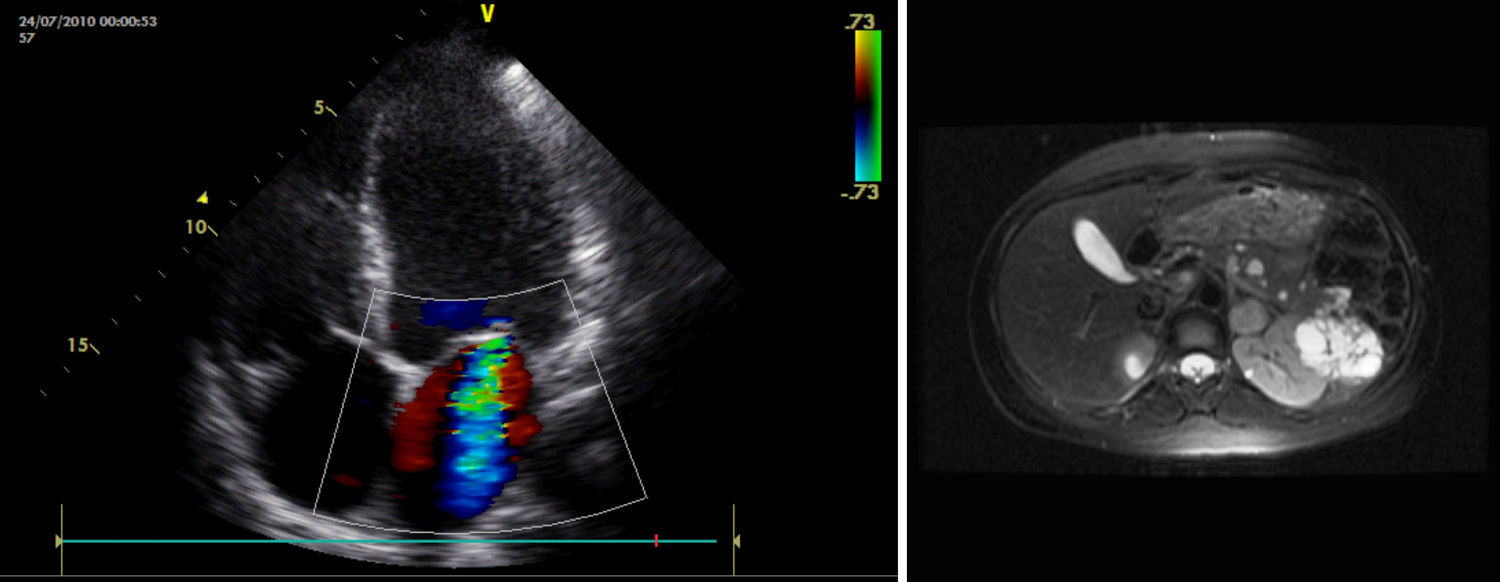
En el electrocardiograma se objetiva taquicardia sinusal a 120 lpm, sin alteraciones en la conducción auriculoventricular ni intraventricular, ni alteraciones en la repolarización. En el análisis practicado destaca NTproBNP de 2.948,7pg/ml (Valores normales (VN): 0-125pg/ml) y un CA 12,5 de 180,5 U/ml (VN 0-35 U/ml), ambos marcadores de congestión y de conocido valor pronóstico en la insuficiencia cardíaca. No se encuentran alteraciones destacables en el resto de parámetros estudiados. La función tiroidea, el perfil lipídico y los marcadores de autoinmunidad son normales. En la radiografía de tórax se observa una silueta cardiaca de tamaño normal con un patrón reticular bilateral sugerente de edema intersticial. Tras los hallazgos descritos, se realiza una ecocardiografía transtorácica, en la que destaca una ligera dilatación ventricular izquierda con FEVI (Fracción de eyección del ventrículo izquierdo) moderadamente deprimida (45%) (VN para su edad 60%) y una insuficiencia mitral grave grado IV/IV con válvula de aspecto normal (fig. 1A).
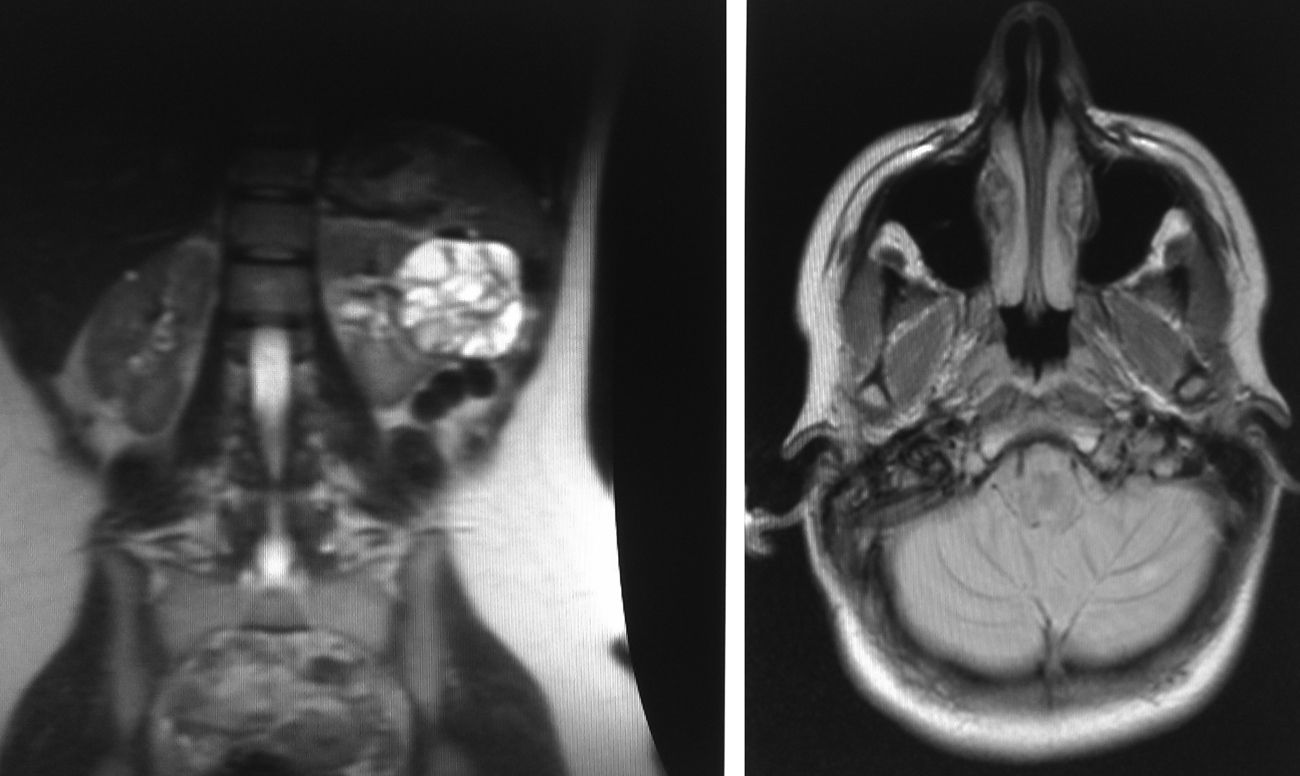
 RM abdominal axial. Tumoración quística heterogénea en riñón izquierdo, de 7,6×6,2cm de diámetro. 2B) RM cerebral. Lesiones hipercaptantes, situadas en el hemisferio cerebeloso izquierdo, una de unos 5mm y la otra más periférica de 3mm, que corresponden a pequeños hemangioblastomas cerebelosos.")
2A) RM abdominal axial. Tumoración quística heterogénea en riñón izquierdo, de 7,6×6,2cm de diámetro.
2B) RM cerebral. Lesiones hipercaptantes, situadas en el hemisferio cerebeloso izquierdo, una de unos 5mm y la otra más periférica de 3mm, que corresponden a pequeños hemangioblastomas cerebelosos.
Ante el diagnóstico de insuficiencia mitral de etiología desconocida, ingresa en el servicio de Cardiología, iniciándose tratamiento con betabloqueante y diurético. En las primeras 48 horas de hospitalización, presenta ligera mejoría de la disnea, con mayor tolerancia al decúbito. Posteriormente, la paciente presenta crisis de ansiedad con sudoración profusa y palpitaciones realizándose electrocardiograma que muestra taquicardia sinusal a 135 lpm. Dado que se trata del primer episodio clínico de insuficiencia mitral, en paciente joven y con mejoría de la clínica, se sospecha origen secundario, solicitándose análisis con autoinmunidad y catecolaminas en orina que son positivas (ácido vanil nandélico 17,5mg/24h (VN 2,9-11,0mg/24h), metanefrinas totales 3012,5μg/24h (VN 0-1000μg/24h), normetanefrina 2987,5μg/24h (VN 105-354μg/24h), catecolaminas totales 657,5μg/24h (VN 217-575μg/24h), noradrenalina 467,5μg/24h (VN 23-105μg/24h), dopamina 177,5μg/24h (VN 190-450μg/24h).
Estos resultados indican la presencia de feocromocitoma y justifican el cuadro clínico. Con este diagnóstico de sospecha en contexto de síndrome de von Hippel Lindau, la paciente pasa a cargo de Servicio de Endocrinología. Se completa el estudio solicitándose RM abdominal y RM cerebral. En la primera, se objetiva una masa suprarrenal izquierda de 2,3cm de diámetro bien delimitada sugestiva de feocromocitoma (fig. 1B). Del mismo modo se informa de una tumoración quística heterogénea en riñón izquierdo, de 7,6×6,2cm de diámetro, en la glándula pancreática se aprecian numerosas formaciones con señal quística tanto en cabeza, cuello como en cola (fig. 2A). La RM cerebral muestra dos pequeñas lesiones hipercaptantes, situadas en el hemisferio cerebeloso izquierdo, una de unos 5mm y la otra más periférica de 3mm, que corresponden a hemangioblastomas cerebelosos (fig. 2B). Una vez realizado el diagnóstico de imagen se contacta con servicio de Urología para planear el abordaje quirúrgico. Se inicia la preparación preoperatoria con dosis crecientes de fenoxibenzamina hasta alcanzar los 20mg/12h. Posteriormente, la paciente presenta un cuadro de palpitaciones, objetivándose taquicardia sinusal que se controla con propranolol a dosis de 20mg/12h. Una vez alcanzada la estabilidad clínica se decide la intervención quirúrgica. Es intervenida 20 días después practicándose una nefrectomía radical y suprarrenalectomia por vía laparoscópica. La paciente no presenta complicaciones inmediatas, objetivándose una mejoría de las cifras tensionales, así como de las palpitaciones. El estudio anatomopatológico mostró un feocromocitoma y un carcinoma renal de células claras. Se solicita un estudio bioquímico posquirúrgico en el que se objetivó la normalización de las catecolaminas y metranefrinas en orina. Destacar que la paciente no presenta antecedentes familiares y que el análisis genético mediante MLPA (Multiplex Ligation-dependent Probe Amplification) de ADN leucocitatrio mostró que la paciente es portadora de una deleción del exon 3 del gen VHL en heterocigosis, no existiendo dudas sobre su predisposición al síndrome de VHL. Se concluye, dado que los familiares no están afectos, que es una mutación de novo. Posteriormente ingresó en Neurología por hipoestesia en hemicuerpo derecho siendo diagnosticada de hemangioblastoma intramedular (febrero 2011), en seguimiento por neurocirugía. Actualmente, la paciente se encuentra asintomática desde el punto de vista cardiovascular realizándose ecocardiografía de control en la que se objetiva ausencia de insuficiencia mitral y FEVI normal.
Ecocardiografía transtorácica (plano apical 4C). Insuficiencia mitral. 1B) RM abdominal corte transversal. Masa suprarrenal izquierda de 2,3cm de diámetro bien delimitada sugestiva de feocromocitoma (señalada con flecha roja).")
El síndrome de VHL es una enfermedad de origen genético de herencia autosómica dominante. El gen afectado es el VHL que se encuentra en el brazo corto de cromosoma 3 (3p25-26)1. Solo un 20% de casos con VHL es familiar, por tanto, existe gran número de mutaciones de novo. En cuanto a las manifestaciones de la enfermedad se incluye la presencia de hemangiomas retinianos (50%), cerebelosos y medulares (30%), carcinoma de células renales (70%), feocromocitoma, tumores pancreáticos, policitemia (15%), etc. En este caso, la paciente presentaba como manifestaciones del síndrome: angiomas retinianos múltiples, feocromocitoma, carcinoma de células renales, hemangioblastomas cerebelosos y medulares. El feocromocitoma en el VHL tiene una presentación diferente que en el feocromocitoma aislado. Para el despistaje del mismo, se recomienda hacer pruebas bioquímicas anuales desde los 10 años a todo sujeto afecto de VHL. Éste aparece con una frecuencia aproximada del 10-20% según los estudios publicados. Es importante incidir en que hasta en la mitad de los casos será de carácter bilateral, aunque estos son raramente malignos. En cuanto a su presentación clínica, hay que destacar que hasta un tercio de los mismos se diagnostica en fase asintomática. Esto se explica por el seguimiento que se realiza a estos pacientes y porque son tumores con menor producción de catecolaminas que los feocromocitomas esporádicos2. La afectación cardíaca puede ser la principal manifestación clínica del feocromocitoma en forma de miocardiopatía catecolamínica, debida al efecto tóxico de las catecolaminas y sus productos de oxidación sobre el miocardio, de forma directa o mediado por el receptor beta adrenérgico3. Las características clínicas de la miocardiopatía relacionada con el feocromocitoma incluyen: hipertensión arterial, cardiomiopatía hipertrófica o dilatada, insuficiencia cardíaca (por efecto directo de las catecolaminas en los pulmones que aumentan la permeabilidad capilar pulmonar, el tono venoso pulmonar y producen daño en el endotelio capilar pulmonar), aturdimiento miocárdico, alteraciones en el electrocardiograma, arritmias cardíacas y paro cardíaco4. El pronóstico de la cardiomiopatía catecolamínica asociada a feocromocitoma depende de la identificación precoz y del tratamiento. El reconocimiento de la cardiomiopatía por catecolaminas debida a feocromocitoma es importante, ya que su tratamiento difiere de otras formas de insuficiencia cardíaca, presentando mala tolerancia a los betabloqueantes A este aspecto, hay que añadir que en la mayoría de casos, tras el tratamiento, el miocardio vuelve a la normalidad en unos meses. El tratamiento de elección del feocromocitoma es la cirugía, previo bloqueo alfa-adrenérgico, como sucedió en el caso presentado5.
