


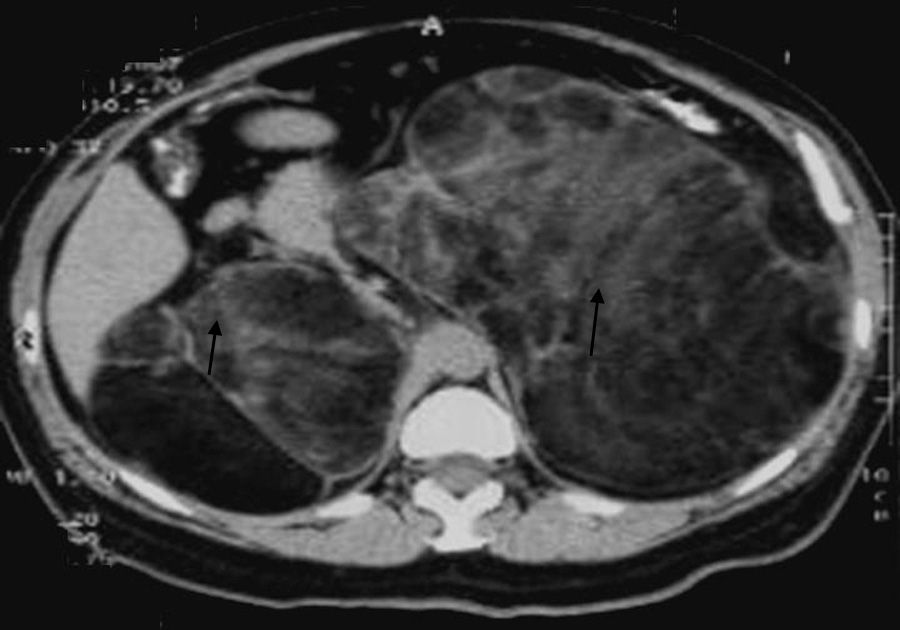
We report the case of a 64-year-old woman who was referred for the evaluation of abdominal pain and bloating over the previous year. She was 1.43m tall and weighed 63kg (body mass index 30.88kg/m2), and her blood pressure was 130/60mmHg. A physical examination revealed abdominal hirsutism, but no hyperpigmentation. Abdominal ultrasound showed several retroperitoneal tumors, and computed tomography (CT) revealed giant adrenal tumors (Fig. 1). She was scheduled for the excision of both adrenal glands by two-stage open surgery. Histopathological examination was diagnostic for bilateral adrenal myelolipomas: the left one measured 21cm×18cm×7cm, and the right one 18cm×11cm×10cm. Oral administration of hydrocortisone and fludrocortisone was started after the second adrenalectomy. Regarding her past medical history, genital ambiguity had been remarked at birth, which determined the diagnosis of congenital adrenal hyperplasia by a pediatric endocrinologist. She underwent two different reconstruction surgeries during childhood. She was prescribed oral glucocorticoids until she was 24 years old; at that time, she deliberately stopped all medication and she discontinued all follow-up visits. She remained asymptomatic for a period of 20 years until the current episode.

The first issue to be considered is the diagnosis of congenital adrenal hyperplasia itself. Unfortunately, specific evaluation by an endocrinologist was not sought until after the completion of adrenalectomy, so hormonal values prior to surgery were not available. The clinical picture and her past medical history suggested the possibility of two types of enzyme blockade. On one hand, she could have been suffering from 11-hydroxilase deficiency. This is characterized by androgen excess and virilization in women, and is not generally associated with acute episodes of adrenal insufficiency because of the elevation of 11-desoxicortisone, which, in turn, leads to hypertension in around 60% of patients. In the case that we present, the patient had never suffered from an acute episode of adrenal insufficiency. However, the low incidence of this enzyme deficiency (less than 1 in 100,000 cases) and the absence of hypertension argued against the diagnosis of this type of blockade.1 The second possible alteration to be considered is 21-hydroxilase deficiency, in its virilizing and non-classical form (no salt loss). Again, virilization of women occurs. Mineral corticoid deficiency may not be present due to sufficient aldosterone production. Currently, the only definitive diagnosis of this syndrome is the identification of gene mutations in cytochromes P450 11 and 21. In patients who are homocygotic for the simple virilizing form of 21-hydroxilase-deficiency, the prevalence of adrenal incidentalomas has been estimated to be around 80%, presumably due to chronic stimulation by corticotropin hormone (ACTH).2 Regarding myelolipomas, these are relatively rare tumors (approximately 1% of all adrenal masses), which consist of mature adipose and hematologic tissues, and are believed to originate from poorly differentiated stromal cells. In the majority of cases, they are asymptomatic and are found as incidentalomas.3–5 Up to now, around 18 cases of 21-hydroxilase deficiency associated with giant myelolipomas have been published, and the diagnosis in most of them was made during adulthood.6,7 The physiopathologic mechanism involved has not been fully elucidated. One hypothesis that could be suggested in this case is that it was the chronic absence of glucocorticoid treatment which led to an elevation of ACTH levels. Studies in animal models have observed an increase in hematopoiesis in adrenal glands secondary to the administration of ACTH. Nevertheless, in previous cases, the expression of ACTH- or androgen-receptors in the tumor itself could not be proven,8 so that the demonstration of this specific pathophysiologic mechanism is difficult to establish. The absence of steroid treatment over a long period of time could have triggered this phenomenon in our patient, as well as the increased prevalence of adrenal tumors in patients with enzyme deficiencies.
In conclusion, adrenal enzyme blockades are associated with an increased prevalence of adrenal tumors. Adrenal myelolipomas are not an exception, since there is not a sole mechanism that explains this pathophysiological association.
Please cite this article as: Garduño-García JJ, Arjona Villicana R, Pimentel L, Pérez Díaz I, Gómez-Pérez FJ. Mielolipoma adrenal gigante de aparición tardía en una paciente con hiperplasia adrenal congénita. Endocrinol Nutr. 2013;60:e33–e34.
