


Hashimoto's thyroiditis (HT) and Graves’ disease (GD) are two very common organ-specific autoimmune diseases which are characterized by circulating antibodies and lymphocyte infiltration. Although humoral and cellular mechanisms have been classically considered separately in the pathogenesis of autoimmune thyroid diseases (AITD), recent research suggests a close reciprocal relationship between these two immune pathways. Several B- and T-cell activation pathways through antigen-presenting cells (APCs) and cytokine production lead to specific differentiation of T helper (Th) and T regulatory (Treg) cells. This review will focus on the cellular mechanisms involved in the pathogenesis of AITD. Specifically, it will provide reasons for discarding the traditional simplistic dichotomous view of the T helper type 1 and 2 pathways (Th1/Th2) and will focus on the role of the recently characterized T cells, Treg and Th17 lymphocytes, as well as B lymphocytes and APCs, especially dendritic cells (DCs).
La tiroiditis de Hashimoto y la enfermedad de Graves son 2 enfermedades tiroideas autoinmunitarias muy frecuentes, que se caracterizan por la presencia de autoanticuerpos circulantes e infiltración linfocitaria. Aunque clásicamente se han considerado los mecanismos humorales y celulares de forma independiente, la evidencia actual apoya la existencia de una relación recíproca entre ambas vías de autoinmunidad en la patogenia de la enfermedad tiroidea autoinmune (ETAI). La presentación de antígenos por parte de las células presentadoras de antígenos y su producción específica de citocinas conduce a una diferenciación de células B y T. El objetivo de este trabajo es revisar los mecanismos celulares implicados en la patogenia de la ETAI. En concreto, se argumentan razones para descartar la visión tradicional, simplista y dicotómica de las vías de las células T helper tipo 1 y 2 (Th1/Th2), y se revisan en detalle las células T recientemente caracterizadas, las células T reguladoras (Tregs) y Th17, así como las células B y las células dendríticas (DC).
Hashimoto's thyroiditis (HT) and Graves’ disease (GD) are two very common organ-specific autoimmune diseases which are characterized by the presence of circulating thyroid antibodies and infiltration by autoreactive lymphocytes of the thyroid gland, and occasionally the orbit. In this setting, an immunological overlap with other autoimmune diseases and a family history, mainly in females, are frequently found. It has been traditionally thought that HT is mainly mediated by a cellular autoimmune response, with a strong inflammatory infiltrate, which leads to destruction and resultant failure to function of the thyroid gland. On the other hand, GD has mainly been considered to be mediated by a humoral autoimmune response, mainly due to the presence of autoantibodies directed against the thyrotropin receptor (TRAb) which stimulate the growth and function of thyroid follicular cells (TFCs), thus leading to development of goiter and hyperthyroidism. However, as in other autoimmune disorders, humoral and cellular immune mechanisms are closely related and cross-linked in AITD and, once they are triggered, they undergo subsequent feedback circuits which reciprocally amplify and perpetuate one of the responses, while inhibiting the opposite, thus denoting the complex mechanisms involved in the pathogenesis of AITD.1,2 Moreover, both immune responses have also been reported in the pathogenesis of Graves’ orbitopathy, one of the most common extrathyroid manifestations of AITD, which may occur in up to 25% of patients with GD.3
Activation of specific pathways for T cell differentiation may depend on the concentration and type of antigen exposure, the nature of the initial antigen-presenting cells (APCs) and, presumably, on still undefined genetic and environmental factors. In this regard, development of experimental models has allowed for increasing understanding of the pathogenesis of AITD. However, a complete understanding of how the complex interaction between genetic susceptibility and environmental factors operates has not been achieved, and further research is needed on why this autoimmune process starts, leading to failure of immunological tolerance at multiple levels.
This review will focus on the cellular mechanisms involved in AITD pathogenesis, specifically the role of the recently characterized T cells, T regulatory (Treg) and T helper (Th) lymphocytes, and also of B lymphocytes and APCs, especially dendritic cells (DCs).
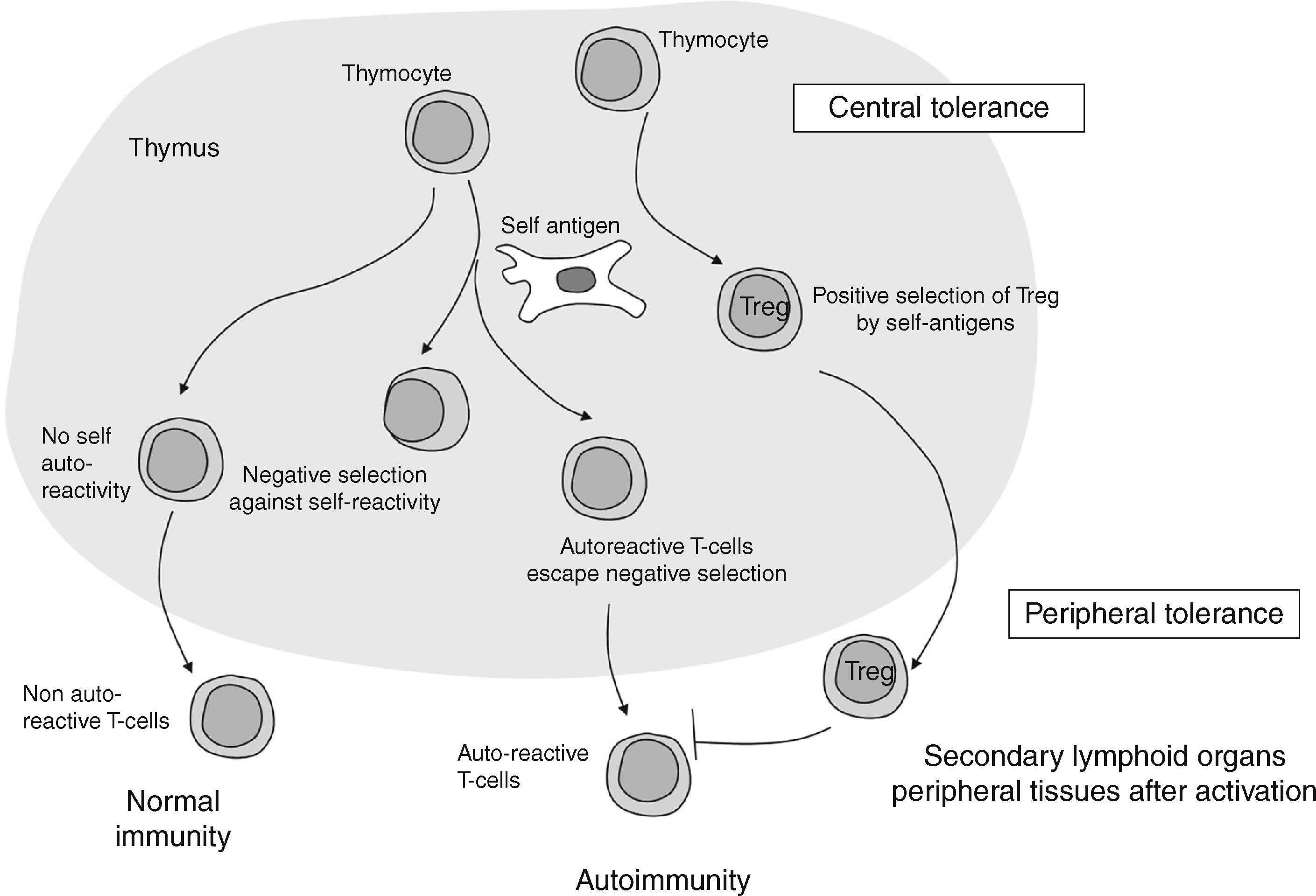
Immune cellular mechanisms in autoimmune diseases: where does it all start?Maturation of T helper CD4+ lymphocytes occurs in the thymus gland after activation on exposure to specific antigens and cytokines. Normally, activation and proliferation of T cells reacting to self-antigens are previously deleted in the thymus by mechanisms of immune central tolerance. In some individuals, however, autoreactive T cells escape from the controlling immune regulatory mechanisms and may activate, proliferate and differentiate, leading to development of an autoimmune response (Fig. 1).

Differentiated T cells will emerge from the thymus to peripheral lymphoid tissue as mature naïve T cells. Once at the periphery, signals based on response to antigens, co-stimulators and/or specific cytokines result in either activation or downregulation of T cells, which results in different subsets of effector cells (Th1, Th2, and Th17) and a smaller population of Treg.4 Each specific lymphocyte subtype will have specific markers which will serve as potential identifiers, and each cell type will subsequently secrete specific cytokines, contributing to fulfillment of their specific actions, as will later be explained. In addition, B-cells will be stimulated to produce autoantibodies, while CD4+ T cells will be the major type of lymphocyte infiltrating the specific tissue/organ. We will further discuss these mechanisms and their specific involvement in AITD.
Main actors in thyroid cellular autoimmunityAntigen-presenting cells and lymphocyte migration to the thyroid glandAPCs, especially DCs, are bone marrow-derived cells of both lymphoid and myeloid stem cell origin that populate all lymphoid organs, as well as almost all non-lymphoid tissues and organs. Human peripheral blood DCs have been classified as conventional DCs (cDCs) and plasmacytoid DCs (pDCs) depending on their specific characteristics.5 As part of the innate immune system, DCs can rapidly respond to environmental damage and mount a primary immune response, mainly thanks to their potent antigen-presenting capacity for stimulating naïve, memory, and effector T cells5. In fact, in order to perform their functions, DCs induce energy and apoptosis of effector lymphocytes, generate Treg cells, and synthesize several cytokines which modulate activation of Treg and effector lymphocytes.5,6 Thus, DCs are functionally classified as immunogenic DCs or tolerogenic DCs when they activate autoreactive T-cells or Treg cells respectively,7 leading to induction or suppression of autoimmune response respectively. Suppression mainly occurs through intrathymic deletion of autoreactive lymphocytes and maintenance of peripheral tolerance to self-antigens.8
Evidence on the role of DCs in the pathogenesis of thyroid diseases was initially found in animals spontaneously developing autoimmune thyroid disease, in which DCs were already seen in the thyroid gland in the initial disease stages. This was followed by a large accumulation of DCs, together with B and T cells.9 However, the role of DCs in thyroiditis induction and development was found to be much more complicated than initially thought.10
Surprisingly, there are relatively few studies available on the role of DCs in human AITD. Several studies have shown that DCs are increased in thyroid infiltrating cells in both GD and HT11–14 and, in this setting, DCs can present immunogenic thyroid-related epitopes (e.g. thyroglobulin) to T cells. Moreover, in a recent quantitative and phenotypic analysis of DCs in patients with AITD, lower numbers of peripheral blood pDCs and a defective expression and/or function of several immunoregulatory molecules were found, suggesting that the altered proportion and function of tolerogenic DCs may contribute to pathogenesis of AITD.15,16 Further research is still needed to better understand all processes where DCs may be involved, especially because recent research has suggested that a potential complex regulation loop between tolerogenic DCs and Treg lymphocytes may also exist.14,17–19
In addition to DCs, TFCs may also act as antigen-presenting cells. The first description of MHC class II molecule expression by thyroid cells20 has been followed by reporting of many other communication pathways between TFCs and infiltrating lymphocytes.4
Moreover, several studies have documented lymphocyte migration to the thyroid gland in AITD following immunogenic thyroid-related epitopes. A critical element in lymphocyte accumulation in the thyroid gland in AITD is expression of selectins and integrins on the endothelium.21 Some β1-integrins, which mediate cell attachment to extracellular matrix proteins, and vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), and E- and P-selectins,22 which are involved in endothelium attachment, are upregulated on endothelial cells in AITD. In addition, some cytokines and chemokines which are able to direct leukocyte migration are also overexpressed in the thyroid in AITD. In this regard, TFCs are able to secrete an array of cytokines, including chemokines, which facilitate localization of the infiltrate in the thyroid gland, acting mostly through activation of the CCR5 and CXCR3 receptors. Infiltrating T cells have an increased interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) production, which stimulates in turn chemokine secretion from TFCs, amplifies the feedback loop, and perpetuates the autoimmune process.23,24
Interestingly, the increased intrathyroid vasculature found in AITD is associated to an increase in circulating vascular endothelial growth factor (VEGF)25 and in the angiopoetin/Tie-2 system. Thus, blood vessels, inflammatory cells, and TFCs may play a significant role in recruitment of mononuclear cells to the thyroid gland and contribute to the pathogenesis of tissue damage seen in AITD.26
B lymphocytesAlthough B lymphocytes are mainly involved in humoral immune mechanisms, their role as a cell itself should not be underestimated. In fact, B cells are activated in patients with AITD. Specifically, B cells play an essential role in development of GD through production of pathognomonic activating autoantibodies (TRAb) against the thyroid-stimulating hormone receptor (TSHR), which leads to increased production and secretion of thyroid hormones. Also, although apparently to a lesser extent, B cells play a pathogenic role in development of HT through production of autoantibodies to the thyroid self-antigens thyroglobulin (Tg, AbTg) and thyroid peroxidase (TPO, AbTPO).27
B lymphocytes recognize intact soluble antigens through their specific cell membrane receptor (an immunoglobulin), synthesize specific antibodies, and present fragments of these antigens to CD4+ T cells. T helper cells will also reciprocally maintain activation of B cells. Particular emphasis has been placed on sequencing thyroid autoantigens and defining B cell epitopes on the TSHR to be able to understand why this receptor is triggered and causes GD.28 However, the usually slow pace of the autoimmune response in AITD entails its spreading and diversification, involving many different B and T cells whose polyclonality exceeds any evidence of a dominant clone, making any attempt to stop these reactions difficult to achieve.
B cells are part of the thyroid lymphocytic infiltrate, and they exert their antibody synthesis activity in the thyroid gland, which shows the significant role of the thyroid itself in promoting AITD persistence. Additional evidence of this finding comes from studies where thyroid autoimmunity was objectively reduced after anti-thyroid treatment or thyroid ablation with radioiodine or surgery. In addition, the beneficial effect of the B cell-depleting antibody rituximab in a number of autoimmune diseases, including Graves’ ophthalmopathy, suggests a critical role of B cell involvement.29 Moreover, recent studies have also suggested the presence of immunoregulatory B cells (Bregs), which secrete interleukin-10 (IL-10), transforming growth factor-β (TGF-β), Fas ligand, and TNF-related apoptosis inducing ligand (TRAIL), which may contribute to maintenance of peripheral tolerance and inhibition of immune responses to specific self-antigens.30
T lymphocytesT lymphocytes may be categorized based on their main role into T cytotoxic (Tc) or T helper (Th) cells. Tc cells infiltrate the thyroid gland and can mediate TFC apoptosis and destruction through activation of the CD95 membrane receptor (Fas-FasL). T helper cells, on the other hand, interact with other cells, including B cells and APCs, and exert their action by synthesizing specific regulatory cytokines. While a majority of Tc cells are CD8+, most Th cells express CD4 on their membranes.
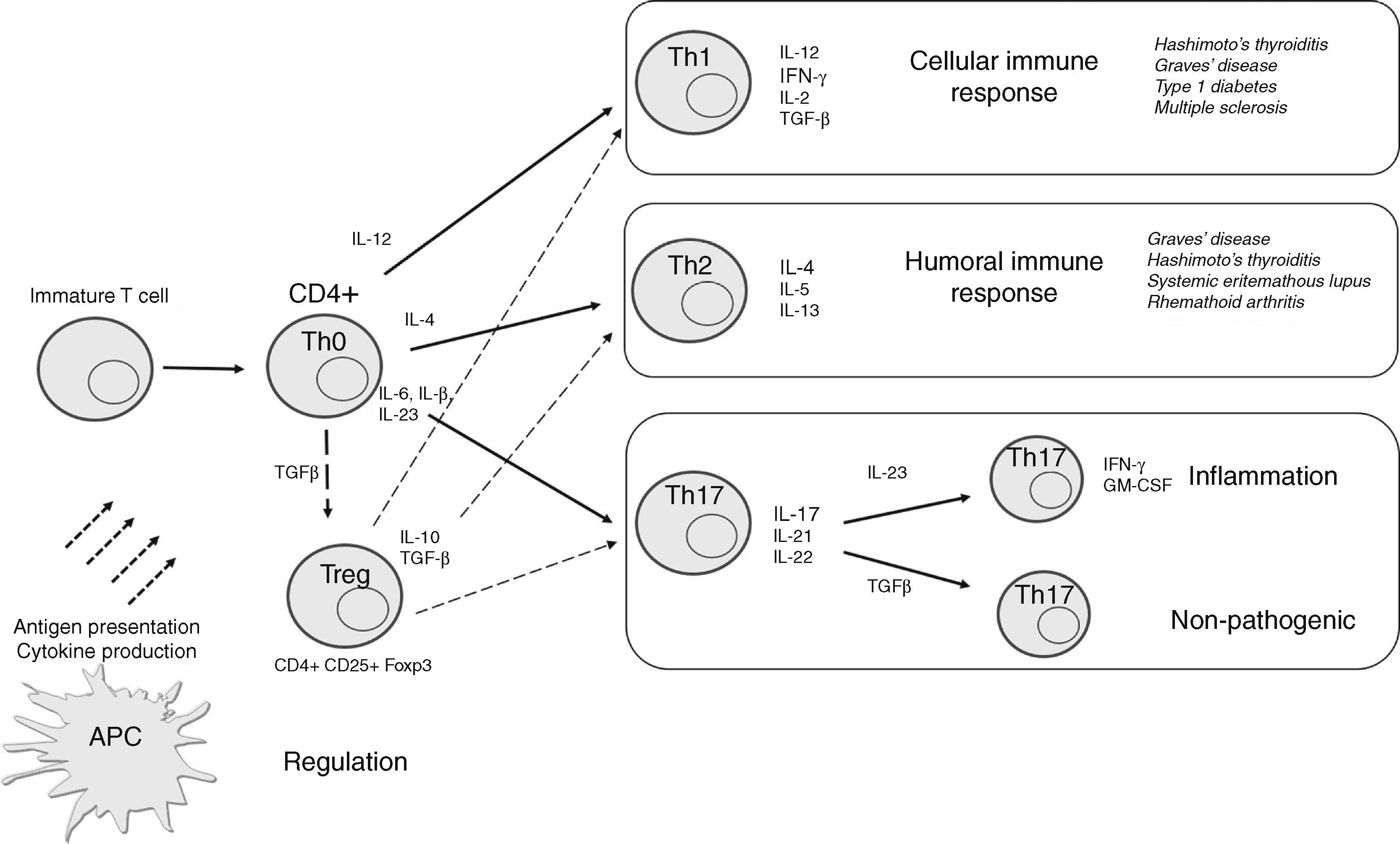
As mentioned previously, Th cells have traditionally been categorized as Th1 or Th2 based on their patterns of cytokine production and effector function. However, some CD4+ T cells may produce both Th1 and Th2 cytokines, and have been called Th0, while other new T cell subsets, including Th17 and Treg cells, have also been identified. In this setting, evaluation of these T cell subsets and their cytokine production in AITD has attracted significant attention in recent years to identify the main mechanisms involved in thyroid autoimmunity, and to develop potential disease outcome markers and treatment strategies. We will review each of these T cell subtypes in AITD (Fig. 2).
, recruited Th1 lymphocytes secrete IFN-γ and TNF-α, which in turn stimulate CXCL10, and create an amplification feedback loop that initiates and perpetuates the autoimmune process, mainly through cellular mechanisms. Th2 cells are mainly involved in humoral immune response. Th17 cells may further develop into pathogenic or non-pathogenic cells depending on their specific stimulation and action. Treg cells mediate an immunosuppressive effect for other T cells.")
Schematic representation of differentiation of CD4+ T cells into specific T cell subsets depending on the cytokines to which they are exposed and their main effect. APCs may present antigens to T cells and mediate their differentiation through synthesis of specific cytokines. In thyroid tissue (and orbital tissue of patients with GO), recruited Th1 lymphocytes secrete IFN-γ and TNF-α, which in turn stimulate CXCL10, and create an amplification feedback loop that initiates and perpetuates the autoimmune process, mainly through cellular mechanisms. Th2 cells are mainly involved in humoral immune response. Th17 cells may further develop into pathogenic or non-pathogenic cells depending on their specific stimulation and action. Treg cells mediate an immunosuppressive effect for other T cells.
Stimulation of naïve CD4+ T cells by IL-12, IFN-γ, IL-2, and expression of the transcription factor T-bet induces their differentiation into Th1 cells. This subset will mainly synthesize IL-1, IL-2, IFN-γ, and TGF-β through activation of the CCR5 and CXCR3 receptors, and will activate cell-mediated immune responses, mainly through cooperation with macrophages and other T lymphocytes.
This response has been the one traditionally reported in patients with HT, where Th1 lymphocytes trigger a strong lymphocyte inflammatory infiltrate of the thyroid which results in subsequent thyroiditis and thyroid gland destruction. Surprisingly, malfunction of this subtype of lymphocytes has also been reported in GD, which rules out the simplistic view that HT was caused by Th1-driven cellular immune mechanisms and GD by humoral Th2-driven mechanisms only. In fact, a recent study noted that T-bet and IFN-γ mRNA expression levels, characteristically Th1-mediated, and IFN-γ plasma levels were significantly upregulated in patients with GD.31
Generation of the IgG subclasses in humans (IgG1, -2, -3, and -4) is also cytokine-biased according to the Th1 or Th2 pathways. In this regard, Th1 cytokines (IFN-γ) drive generation of the IgG1 subclass, which is involved in the early stage of humoral response. In the specific setting of GD, TRAb activity is mostly confined to the IgG1 fraction,32,33 although a paradoxical mixed picture may sometimes exist: IgG1, IgG4, and both.34 Thus, overall, the very strong bias toward IgG1 subclass TSHR autoantibodies suggests that GD is also a Th1- (not only Th2) associated disorder.1
Th2 responsePresence of IL-4 inhibits differentiation of naïve CD4+ T cells into Th1 cells and favors generation of Th2 lymphocytes, which mainly synthesize IL-4, IL-5, IL-6, IL-10, and IL-13. Several experimental models have shown that Th2 cells mainly interact with B and plasma cells, leading to increased production of antibodies which will mediate a humoral immune response.1,4 Specifically, Th2 cytokines (mainly IL-4) drive IgG4 production, which is associated to prolonged immunization.
As mentioned above, it was traditionally thought that this Th2 response was the predominant mechanism involved in GD, in which persistent increased levels of autoantibodies directed against TSHR stimulate the growth and function of thyroid follicular cells, thus leading to development of goiter and hyperthyroidism. Once again, however, this dichotomy is not so simple and straightforward. In fact, although HT is characterized by cell-mediated damage to thyroid cells, autoantibodies to TPO and Tg are also a key component in its pathogenesis. In this regard, there is evidence that these antibodies may contribute to thyroid destruction by means of antibody-dependent cell-mediated cytotoxicity and complement activation, mainly through IgG1 (not IgG4), which occurs in parallel to T cell cytotoxicity. Thus, viewed from the perspective of thyroid-specific autoantibodies, both HT and GD are Th1- and Th2-associated diseases.1
Treg cellsTreg lymphocytes were first described as CD4+ suppressor cells. Some years later, they were characterized as regulatory cells that expressed the transcription factor Foxp3 and high constitutive levels of the alpha chain of IL-2 receptor (CD25+). These natural CD4+ CD25+ Foxp3+ Treg cells are considered as major components of Treg cells, and emerge from the thymus as fully differentiated cells. Most of these natural Treg cells recognize self-antigens, exhibit a limited proliferation capacity when their antigen receptor is engaged, and inhibit activation, proliferation and cytokine synthesis by conventional lymphocytes. In addition, they are involved in cell–cell contact mechanisms, which contribute to immune response regulation. Moreover, the immunosuppressive effect of Treg cells is complemented by production of several anti-inflammatory cytokines such as TGF-β, IL-10, and IL-35.35 Therefore, Treg cells act as the main contributors to prevent autoimmune responses by maintaining homeostatic mechanisms that suppress autoreactive T cell proliferation and promote autoreactive T cell anergy.36 However, autoreactive T cells escape in some cases from the control of these immunoregulatory processes, thus leading to a complete autoimmune response including activation, proliferation, and differentiation. In this regard, defects in Treg cell number and/or function may contribute to loss of peripheral self-tolerance, leading to development of autoimmune diseases.
Experimental models of mutations in the gene that encodes Foxp3 (“scurfy mice”) have shown systemic activation of T cells, lymphocytic infiltration of tissue, including thyroid tissue, and increased synthesis of chemokines, leading to premature death. Similarly, Foxp3 mutations in humans may lead to development of inflammatory and autoimmune diseases such as IPEX syndrome (an X-linked syndrome which comprises immune dysregulation, polyendocrinopathy, including thyroiditis, and enteropathy).37
In the particular setting of AITD, several reports confirmed defective function of Foxp3+ Treg cells in patients with HT or GD.14,17,38–42 Levels of CD4+ Treg cells may be variable in the peripheral blood from patients with AITD, but a finding that has been more consistent across different reports is an increased proportion of these lymphocytes as the main component of the thyroid tissue infiltrate.38,40,42–44 These lymphocytes are however dysfunctional and unable to effectively downregulate the ongoing autoimmune process and inflammatory phenomenon.14,17,38–42,44
Another subset of Treg cells, characterized by constitutive expression of CD69, has recently been described in mice. These cells are CD4+ CD69+ TGF-β+, but do not express Foxp3 or CD25 and exert an important immunosuppressor effect. Several studies suggest that these lymphocytes may also play a significant role in autoimmune disease in humans, presumably through defective function of the immunosuppressive effect of CD69. In fact, patients with AITD have an increased population of these CD69+ cells in both peripheral blood and thyroid tissue, which are apparently unable to down-modulate the autoimmune response and tissue damage.44
Th17 cellsDifferentiation of Th17 cells is mainly induced by the combined action of IL-β, IL-6, and IL-23, together with expression of the transcription factor retinoic acid receptor-related orphan receptor C2 (RORC2) and activation of the signal transducers and activators of transcription-3 (STAT3) intracellular pathway.45,46
Th17 lymphocytes are mainly characterized by the synthesis of IL-17A, IL-17F, IL-21, and IL-22,47 which contribute to the release of other pro-inflammatory mediators (such as chemokines, TNF-α and IL-β) by stimulating epithelial cells, fibroblasts, and macrophages. Th17 cells may exert a pathogenic role if specific further differentiation occurs.48,49 For instance, sustained exposure to IL-23 results in conversion of classical Th17 lymphocytes into pro-inflammatory, pathogenic cells able to synthesize IFN-γ and granulocyte-macrophage colony-stimulating factor (GM-CSF).50 By contrast, continuous effect of TGF-β on classical Th17 cells induces their conversion into non-pathogenic lymphocytes involved in mechanisms against different extracellular bacteria and fungi and tissue repair48,49,51–53 (Fig. 2). These two different subtypes may be easily differentiated and identified by multiparametric flow cytometry, mainly through detection of specific markers: pathogenic pro-inflammatory Th17 lymphocytes are CD4+ CXCR3+ CD161+ MDR1/CD243+ IFN-γ+IL-17+, and IL-10-, while non-pathogenic cells are CD4+ CXCR3+ CD161- MDR1/CD243- IL-17+, and IL-10+.49
Furthermore, cross-regulation of Th1 and Th17 cells, which would significantly affect control of the number and function of Treg cells, has been reported.54,55 In this regard, pathogenic Th17 lymphocytes may differentiate into Th1-like (non-classic Th1) cells, which synthesize IFN-γ and GM-CSF, but not IL-17, contributing to tissue damage in autoimmune conditions.2 In addition, Th17 cells may evolve into Foxp3+ Treg cells, which may further antagonize the effect of pathogenic Th17 cells. Interestingly, the opposite differentiation route (i.e., from Treg to Th17), as well as a mixed phenotype Th17/Treg, have also been observed.56–58 All these findings denote the great plasticity of Th17 cells and the multiple and complex roles they exert both in healthy conditions and in immune diseases.2 This complex loop between Th17 and Tregs may be determinant in the pathogenesis of AITD, but still deserves further research in this specific setting30,59
A higher proportion of Th17 lymphocytes and elevated levels of Th17 cytokines were found in the peripheral blood and thyroid gland from patients with AITD, mainly those with HT, and peripheral blood lymphocytes from these patients showed increased induction of the RORC2 gene and an enhanced capability to differentiate in vitro into Th17 cells.60 These findings were quite interesting because they suggested a potential relevant role of Th17 cells in the inflammatory phenomenon and tissue destruction seen in HT. In fact, these data were confirmed in subsequent studies,61–63 and overall increased levels of IL-17+ T cells and a decreased Treg/Th17 cell ratio were seen in HT patients.64,65 These IL-17+ cells are likely to correspond to pathogenic pro-inflammatory Th17 lymphocytes (and very few non-pathogenic cells), but their specific phenotype has not been elucidated yet.2 Circulating platelet-derived microvesicles are significantly increased in AITD patients and can inhibit differentiation of Foxp3+ Treg cells and induce differentiation of Th17 cells.66 Cytokines related to Th17 differentiation, including IL-6 and IL-15, were also detected at high levels in sera from patients with HT.60 The reason why IL-6 and IL-15 levels are increased in the specific setting of AITD still needs to be clarified, but such elevated levels may be related to genetic factors, including different polymorphisms.
Other additional molecules, such as leptin,67 glucocorticoid-induced tumor necrosis factor receptor ligand (GITRL),68 and galectins16 have also been reported as potential regulators of the function of Th17 cells in AITD. However, the influence of thyroid hormones on Th17 and Treg still needs to be elucidated.
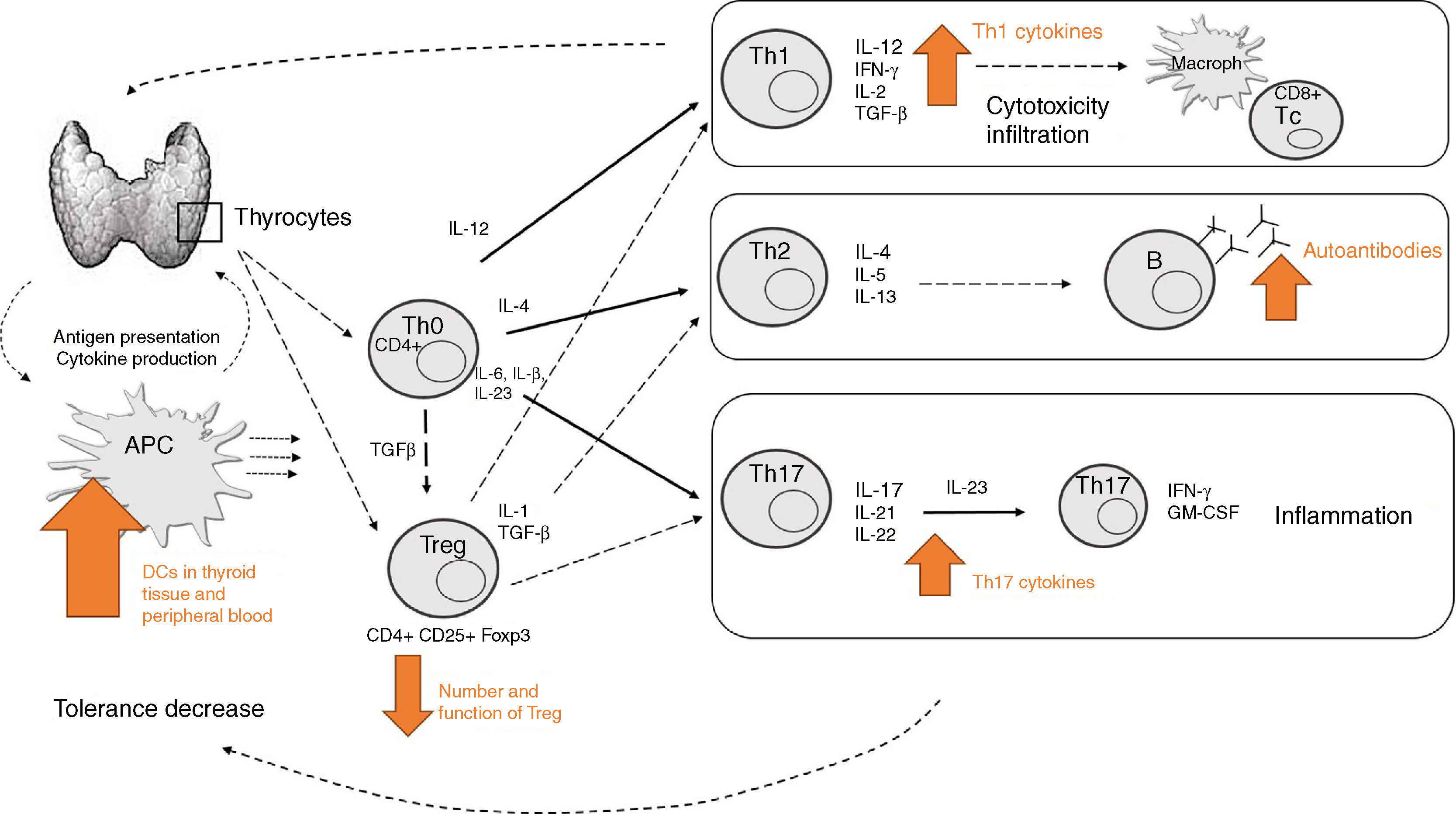
How do all these mechanisms come together in thyroid autoimmune disease?The mechanisms previously described denote the complexity of immune mechanisms involved in AITD development. Fig. 3 represents a simplified summary of the currently known major components. However, there is probably still much to be learned about other potential cell subsets yet to be identified, and their specific functions.
, which leads to specific Th cell differentiation depending on the specific cytokine production. Increased Th1 activity will mediate cytotoxicity and infiltration (mainly through IL-12 and IFN-γ). Increased Th2 activity (mainly mediated through IL-4) will lead to increased auto-antibody production. Increased Th17 activity (mediated through IL-17) will lead to increased differentiation of pathogenic Th17 cells and subsequent inflammation. Decreased Treg activity jeopardizes the immunosuppressive effect for other T effector cells.")
Summary of the main mechanisms involved in development of thyroid autoimmune disease. APCs in thyroid tissue and peripheral blood may recognize self-antigens and present them to T cells (decreased self-tolerance), which leads to specific Th cell differentiation depending on the specific cytokine production. Increased Th1 activity will mediate cytotoxicity and infiltration (mainly through IL-12 and IFN-γ). Increased Th2 activity (mainly mediated through IL-4) will lead to increased auto-antibody production. Increased Th17 activity (mediated through IL-17) will lead to increased differentiation of pathogenic Th17 cells and subsequent inflammation. Decreased Treg activity jeopardizes the immunosuppressive effect for other T effector cells.
Current therapeutic approaches to thyroid disease, including well-known therapies with levothyroxine, anti-thyroid drugs, radioiodine, corticosteroids, or radiotherapy, are usually sufficiently effective for controlling thyroid function, but not always for orbital complications. Understanding of the specific immune mechanisms involved in pathogenesis of AITD could guide us in development of targeted therapies, which would significantly improve treatment efficacy and patient outcome. In this regard, novel biological agents, such as monoclonal antibodies against surface antigens, anti-cytokine or cytokine receptors, and blockers of intracellular signaling pathways, may help us modulate the aberrant immune response seen in AITD and contribute to overall improvement. In this regard, biological therapies, including anti-CD20 rituximab or anti-IL-6 receptor tocilizumab, have been successfully used to treat Graves’ orbitopathy.29,69 Moreover, mediators that antagonize or inhibit the action of Th17 cells, and/or approaches to increase the function and/or number of Treg cells, could be interesting topics for future research. In this way, reintroduction of tolerance or modification of the intrathyroid autoimmune process offer more chances for successful treatment.
ConclusionsAITD results from aberrant immune mechanisms and loss of tolerance of organ-specific self-antigens. In addition to humoral responses, recent research has pointed out the concomitant relevance of immune cellular mechanisms. In this regard, differentiation of CD4+ cells in the particular setting of specific immune mediators (cytokines, chemokines, and other molecules) results in differentiation of various T cell subsets. Assumption of the simple dichotomous view that humoral immunity is driven by Th2 cytokines (such as IL-4), leading to GD, and cellular immunity is driven by Th1 cytokines (such as IFN-γ), leading to HT, has been outweighed by cumulative data showing that AITD, as a whole, comprises elements of both Th1 and Th2 subtypes. In addition, the recently described T cell subtypes Th17 and Treg have also been attributed an essential role in pathogenesis of AITD. In fact, an imbalance has been seen in their number and/or function, and further studies will help us elucidate if they could become a potential target for future effective therapies for AITD.
Conflicts of interestThe authors declare no conflicts of interest.
