


Acromegaly is a condition characterized by chronic growth hormone (GH) hypersecretion due to a pituitary adenoma in more than 95% of cases. Cardiovascular changes, found in up to 60% of patients, represent the main cause of morbidity and mortality. Pituitary apoplexy (PA) is the clinical syndrome that occurs after spontaneous hemorrhage or infarction of the pituitary gland, usually in the setting of a pre-existent adenoma. We report a case of acromegaly diagnosed after a heart failure event in a patient who, after experiencing a PA episode, showed resolution of acromegalic cardiomyopathy (AM) and cure of acromegaly.
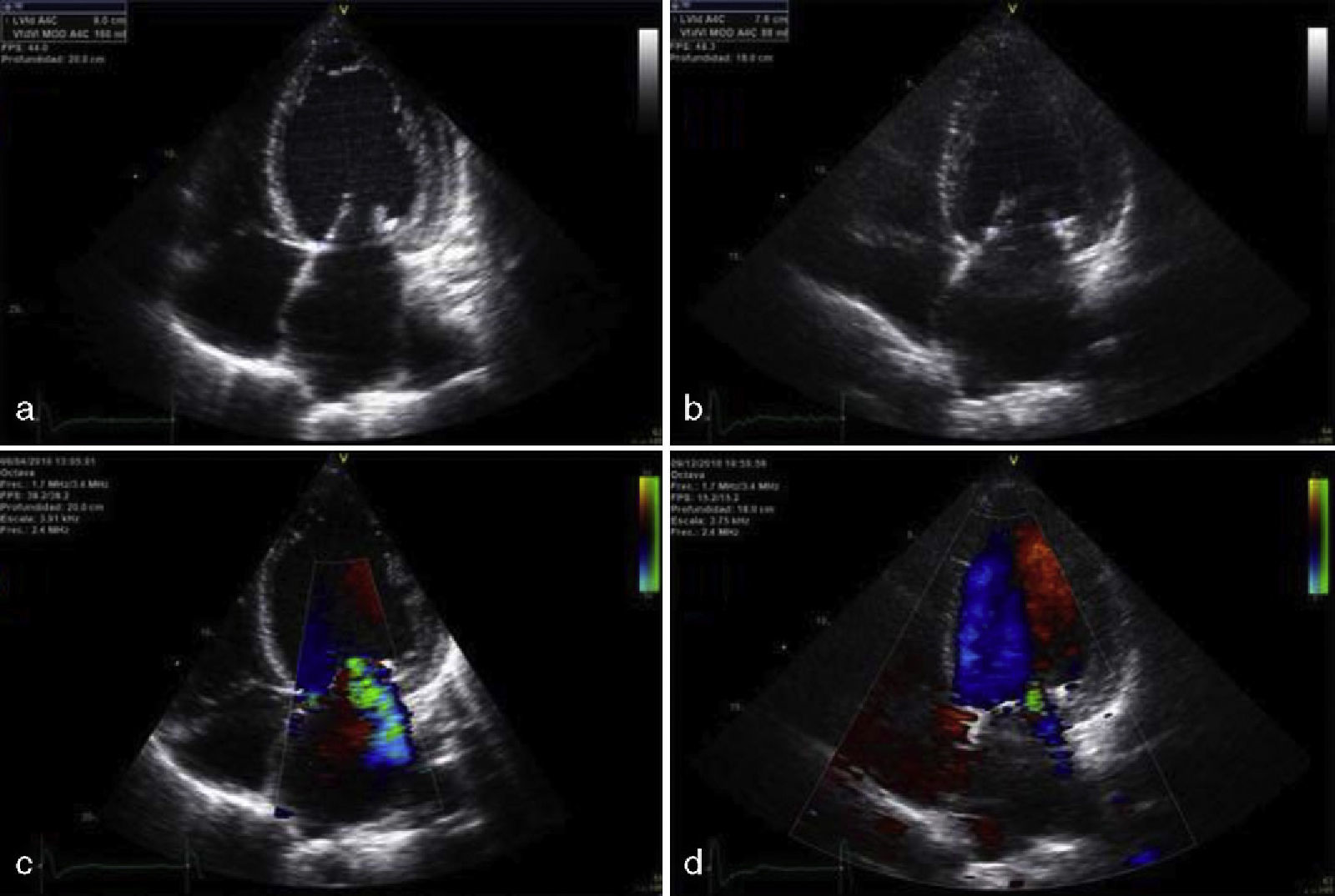
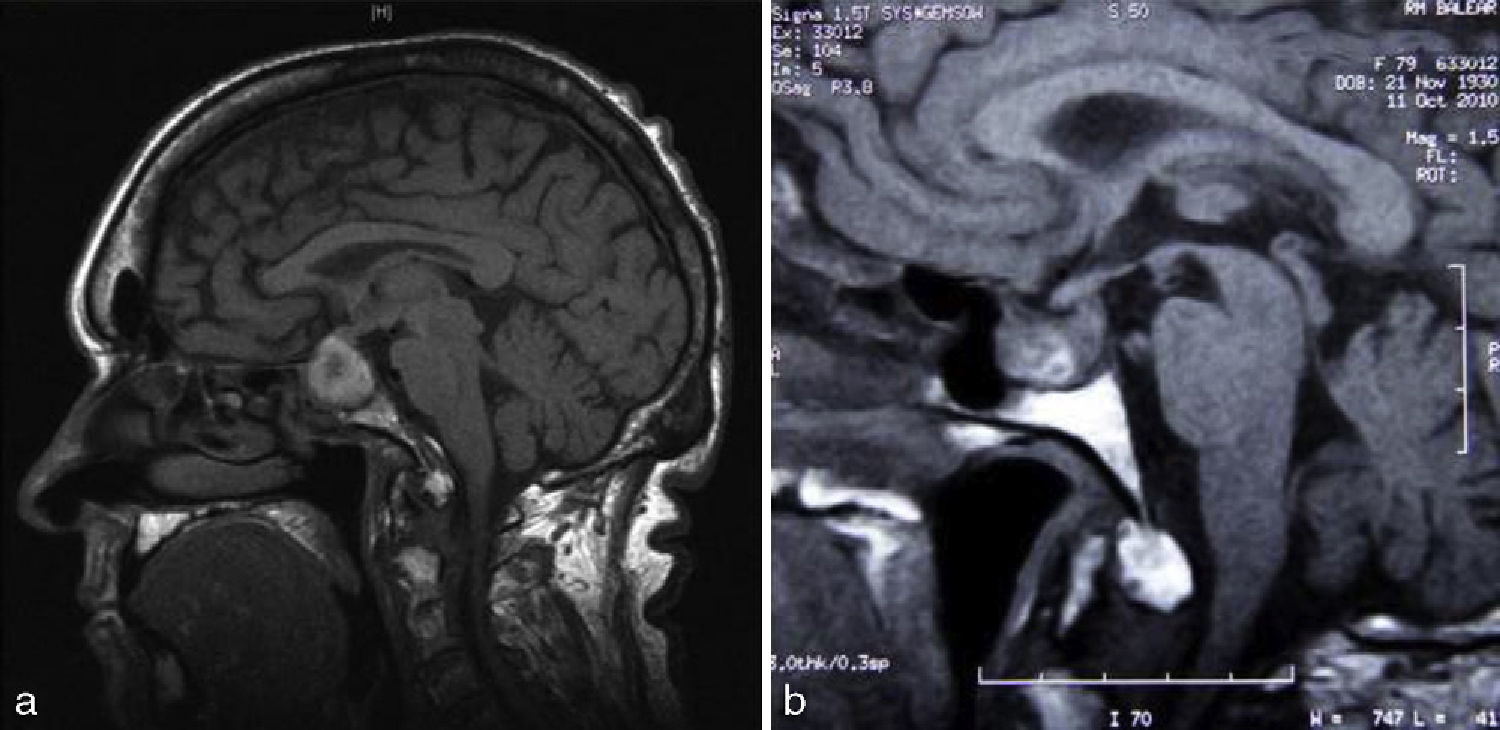
The patient was a 79-year-old woman with a history of high blood pressure (HBP) and type 2 diabetes mellitus (T2DM) diagnosed 15 years before, well controlled and being treated with amlodipine, lisinopril, hydrochlorothiazide, metformin, and glibenclamide. The patient attended the emergency room for anginal chest pain with complete left bundle branch block in ECG and no elevation of myocardial necrosis biomarkers. She was admitted with a diagnosis of acute coronary syndrome, and treatment was started with double antiaggregation therapy (aspirin and clopidogrel) and low molecular weight heparin. The patient experienced rapidly progressive dyspnea, with blood pressure levels of 220/120mmHg, and showed clinical and radiographic signs consistent with acute lung edema. A physical examination showed a weight of 65kg, a height of 158cm (body mass index 26kg/m2), a 3/6 mitral systolic murmur, and coarse facial features, with a growth of acral parts, macroglossia, prognathism, diastema, acrochordons, and goiter, all of them suggesting acromegaly. The patient reported hand growth and a shoe size increase of two sizes over the previous 15 years. Laboratory tests results included glucose, 107mg/dL; glycosylated hemoglobin, 7.9%; cholesterol, 177mg/dL; triglycerides, 64mg/dL. An ECG showed complete left bundle branch block with secondary repolarization changes. Chest X-rays revealed cardiomegaly and alveolar–interstitial infiltrates with a bilateral butterfly pattern. Transthoracic echocardiogram showed a dilated left atrium 56.4mL/m2 in size, a slightly hypertrophic, dilated left ventricle of 70mm with advanced ventricular systolic dysfunction (20% ejection fraction as estimated by the biplane Simpson's method) due to moderate septal hypokinesia and severe hypokinesia of all other segments, grade III/IV mitral regurgitation and grade I/IV tricuspid regurgitation (Fig. 1a). After a favourable course and based on suspicion of AC, hormone tests and magnetic resonance imaging (MRI) of the pituitary gland were requested. The patient experienced an episode of sudden headache associated with nausea, vomiting, decreased visual acuity, and blood pressure of 50/30mmHg with an impaired general condition. Laboratory tests showed low sodium levels of 127mg/dL. PA and secondary adrenal insufficiency were suspected, and intravenous glucocorticoids were therefore started. Pituitary MRI showed a 2.3cm×2.3cm intrasellar lesion with images suggesting hemorrhage contacting with optic chiasma which was consistent with apoplexy of a macroadenoma (Fig. 2a). The campimetry performed was not evaluable. Hormone tests showed hypogonadotropic hypogonadism with follicle-stimulating hormone levels of 0.4mIU/mL (23–116), undetectable luteinizing hormone (5.9–54), and an estradiol level of 12.6pg/mL (0–37); secondary adrenal insufficiency with 1.52μg/dL (4.3–25) of cortisol and 6.9pg/mL (1–46) of corticotropic hormone; prolactin, 11.2ng/mL (1.8–20.3); thyrotropin, 0.07μIU/mL (0.35–5.5), thyroxine unbound to protein, 1.25ng/dL (0.89–1.76) and triiodothyronine unbound to protein, 1.61pg/mL (2.3–4.2), suggesting euthyroid sick syndrome; GH: 1.07ng/mL (0.05–7.4); insulin-like growth factor I (IGF-I), 120ng/mL (55–166). Prior GH and IGF-I levels were not available. The patient was discharged on metformin, enalapril, carvedilol, furosemide, spironolactone, and hydrocortisone. After 8 weeks of follow-up, a transthoracic echocardiogram showed a decreased left ventricular end-diastolic diameter with increased ejection fraction (50% as measured with Simpson's biplane method) and decreased mitral regurgitation (Fig. 1b). Pituitary MRI performed at six months showed a decreased tumor size (1.1cm×1.7cm) with less hemorrhagic content (Fig. 2b). Hypogonadotropic hypogonadism and secondary adrenal insufficiency persisted. The patient had subclinical hyperthyroidism (thyrotropin, 0,03,: thyroxine unbound to protein, 1.54) due to goiter, normal prolactin; GH 0.54ng/mL and IGF-I 40.6ng/mL, glycosylated hemoglobin of 5.6%, and normal campimetry.
 that normalized 8 months after pituitary apoplexy (b), showing an improved ejection fraction. Note the decrease in severity of mitral regurgitation, shown before (c) and after pituitary apoplexy (d).")
Apical four-chamber echocardiographic view showing the left ventricle with increased end-diastolic volume (a) that normalized 8 months after pituitary apoplexy (b), showing an improved ejection fraction. Note the decrease in severity of mitral regurgitation, shown before (c) and after pituitary apoplexy (d).
. Tumor size decrease at 6 months of follow-up (b).")
Morphological and functional changes due to chronic GH and IGF-I excess occur in AC. In the early stage of the disease, the trophic and inotropic effects of GH cause myocardial hypertrophy with biventricular involvement characteristic of AC and hyperdynamic syndrome. AC evolves with more severe ventricular hypertrophy, and interstitial fibrosis leading to diastolic dysfunction and exercise intolerance occurs.1 In the final stage, increased myocardial apoptosis and fibrosis occur leading to ventricular dilation, systolic and diastolic dysfunction. Functional valve impairment may occur, and mitral regurgitation is common.2,3 Patient age and long exposure to excess DG and IGF-I are the main determinants in AC development, occurring in up to 90% of patients.4 Heart failure is seen in 3–10% of patients and is more significant in patients with long-standing acromegaly, HBP, and valve dysfunctions.5 There are a number of factors associated with AC progression such as HBP, T2DM, hypertriglyceridemia, thyrotoxicosis, coronary artery disease, arrhythmia, and respiratory diseases. A differential diagnosis of AC should mainly be made with dilated cardiomyopathy of ischemic, hypertensive, or idiopathic origin. In the reported case, the lack of elevation of myocardial necrosis markers and changes in left ventricular segmental motility made an ischemic origin of cardiomyopathy unlikely. The possibility of a dilated hypertensive heart disease was considered, but the patient had a history of long-standing HBP well controlled on adequate drug treatment. AC usually experiences significant regression, and resolution in some cases, after acromegaly is cured. This occurred in the reported patient, supporting the diagnosis of AC.
The treatment of acromegaly includes transsphenoidal surgery, drug treatment (somatostatin analogues, GH receptor antagonists, dopamine agonists), and radiotherapy.6 Cure after an episode of PA is exceptional. PA occurs in 0.6–10% of pituitary adenomas and its clinical presentation ranges from asymptomatic forms to life-threatening emergencies.7 The most common clinical signs include headache, vomiting, and visual field and cranial nerve impairment. PA may cause any transient or permanent pituitary hormone deficiency. The mechanisms involved have yet to be clearly established. Vascular tumor abnormalities, ischemia due to rapid adenoma growth, and the compression of the superior hypophyseal artery against the diaphragm are mechanisms which contribute to apoplexy. Advanced age may increase pituitary fibrosis and vessel fragility. T2DM may contribute to the occurrence of an infarction due to the degenerative changes it causes in pituitary microcirculation.7,8 Potential triggering factors include stimulation tests with hypothalamic hormones, anticoagulant and antiaggregant treatment, blood pressure changes, meningitis, X-ray contrast agents, surgical procedures, head trauma, and some drugs (atenolol and butformin). The treatment of PA is based on surgical decompression and hormone replacement therapy. The initial approach consists of life support measures, water and electrolyte replacement, laboratory tests to assess hormone deficiencies, and high corticosteroid doses, because secondary adrenal insufficiency is the main cause of morbidity and mortality. Early surgical decompression is recommended if decreased visual acuity, severe persistent visual field deficiency, and impaired consciousness exist. Hormone replacement will depend on associated deficiencies, with hypopituitarism remaining in 50–87% of cases.9,10 Fraser et al. reported 22 cases of remission of acromegaly after pituitary apoplexy. Eight cases occurred in patients with no prior formal diagnosis of acromegaly, with severe retro-orbital headache being the most common symptom. GH normalized in 19 patients. Apoplexy caused hormone deficiency in 15 patients, and improvement or normalization of a prior deficiency in 2 patients. Improvement or cure of T2DM and HBP improvement occurred in 7 patients and 1 patient respectively.7
Our patient had advanced AC due to undiagnosed, long-standing acromegaly, advanced age, and two factors that could have influenced the progression of cardiomyopathy, HBP and T2DM. Double antiaggregation therapy, anticoagulation, and blood pressure changes possibly triggered apoplexy. Secondary adrenal insufficiency and gonadotropin deficiency occurred. PA cured acromegaly which, combined with specific medical treatment (betablockers, angiotensin-converting enzyme inhibitors, aldosterone antagonists), caused the regression and virtual resolution of PA after 8 months of follow-up. An improvement occurred in T2DM, which is currently being treated with metformin. The glycosylated hemoglobin level is 5.6%.
The early diagnosis and treatment of acromegaly, combined with the control of other cardiovascular risk factors, is important in order to decrease associated cardiovascular morbidity and mortality and to improve the quality of life of the patient.
A multidisciplinary approach to acromegalic patients requires an understanding of the symptoms associated with PA, the triggering factors, and the treatment required.
It is essential to monitor patients who have experienced PA for new hormone deficiencies or disease recurrence, which may occur even several years after the episode.
Please cite this article as: Serra Soler G, et al. Resolución de miocardiopatía acromegálica tras apoplejía hipofisaria. Endocrinol Nutr. 2012;59(7):459–61.
