








El linfoma primario de colon es un tumor poco frecuente del tracto gastrointestinal. Con el propósito de informar un caso de linfoma primario de colon y revisar la literatura médica, se presenta este reporte sobre una mujer de 70 años, donde el diagnóstico se estableció mediante el estudio anatomopatológico e inmunohistoquímica de la lesión tumoral, tomada mediante cirugía. El diagnóstico final fue linfoma de células B tipo MALT, de bajo grado (Linfoma no Hodgkin).
Primary colonic lymphomas are very uncommon gastrointestinal tumors. We present the case of a 70-year-old woman. The diagnosis was made by anatomopathology and inmunohistoquimic study of tumor lesion. The patient was subjected to exploratory laparotomy and the final diagnosis was given to be a low grade MALT type Lymphoma B cells (Non-Hodgkin's Lymphoma).
Introducción
Los linfomas gastrointestinales primarios representan una entidad clínico patológica diferente a los linfomas ganglionares, son los más frecuentes entre los linfomas extra ganglionares primarios y constituyen entre el 1% al 4%, de todos los tumores malignos del tubo digestivo.1 El linfoma MALT (Mucosa Associated Lyphoid Tissue o mucosa asociada al tejido linfoide) es un tumor esporádico, que procede de las células B del MALT, siendo el más común del hemisferio occidental. Las características biológicas de estas neoplasias son distintas, comparada con las de los linfomas originados en los ganglios linfáticos, ya que se pueden comportar como tumores focales en sus etapas tempranas, resultando asequibles a la resección quirúrgica. La recurrencia puede afectar exclusivamente al tracto gastrointestinal.2 Los cambios genotípicos son diferentes de los observados, en los linfomas ganglionares. Este tipo de linfoma gastrointestinal suele afectar a individuos adultos, no existe predilección por ningún sexo y se puede originar en cualquier lugar del tracto gastrointestinal: estómago (55% a 60% de los casos), intestino delgado (25% a 30%), colon proximal (10% al 15%), y colon distal (hasta 10%), rara vez suele afectarse el apéndice.3 El diagnóstico de linfoma primario gastrointestinal puede ser sugerido por el cuadro clínico, estudio de imágenes y endoscopía, y definido por estudio anatomopatológico y de inmunohistoquímica.
El objetivo de este artículo, es informar el caso por lo infrecuente que resulta esta patología, y revisar la entidad clínica.
Presentación del caso
Femenina de 70 años de edad, con antecedentes de artritis reumatoide de 20 años de evolución, en tratamiento con metotrexate, leflunomida y prednisona, en los últimos 10 años. Inicia con un cuadro de seis meses de evolución caracterizado por astenia, adinamia, distensión abdominal, cambio en el hábito intestinal con heces disminuidas de consistencia y hematoquezia, así como anorexia, sin otra sintomatología agregada. Veinticuatro horas antes del ingreso, inicia con dolor abdominal generalizado. Al examen físico presenta palidez de tegumentos discreta, masa abdominal dolorosa de 10 cm de diámetro, en fosa y flanco derechos. Se practicaron exámenes de laboratorio que mostraron anemia normocítica normocrómica, con hemoglobina de 11 mg/dL (1.1 mmol/L), química sanguínea sin alteraciones, placa de tórax normal. Se practicó tomografía computarizada de abdomen, que mostró tumoración en colon ascendente de 10 x 8 x 8 cm de diámetro, bordes bien definidos y sólida (Figura 1). El antígeno carcinoembrionario fue normal. Se realizó colonoscopia, en la cual se demostró tumoración en colon ascendente, que protruía hacia la luz de 4 x 4 x 3 cm de diámetro aproximadamente, no obstructivo, de bordes bien definidos (Figura 2), de la cual se tomo biopsia. Las biopsias reportaron solamente infiltrado linfocitario, en el estudio trasoperatorio.
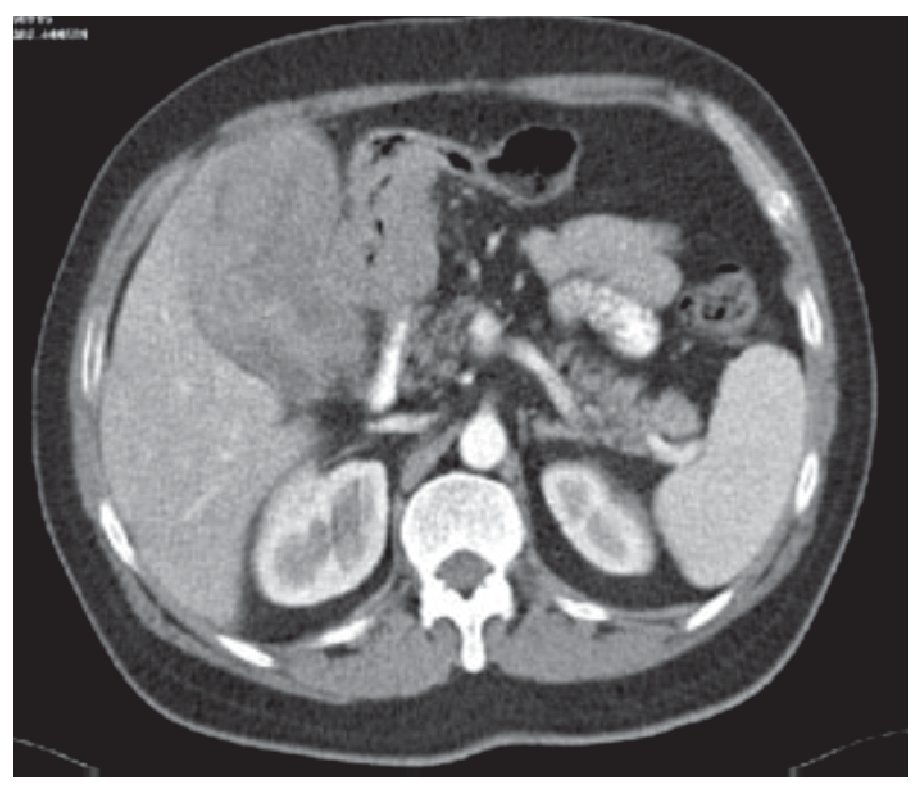
¡ð Figura 1.Tumoración en colon ascendente, de 10 x 8 x 8 cm de diámetro, con bordes bien definidos, sólido.
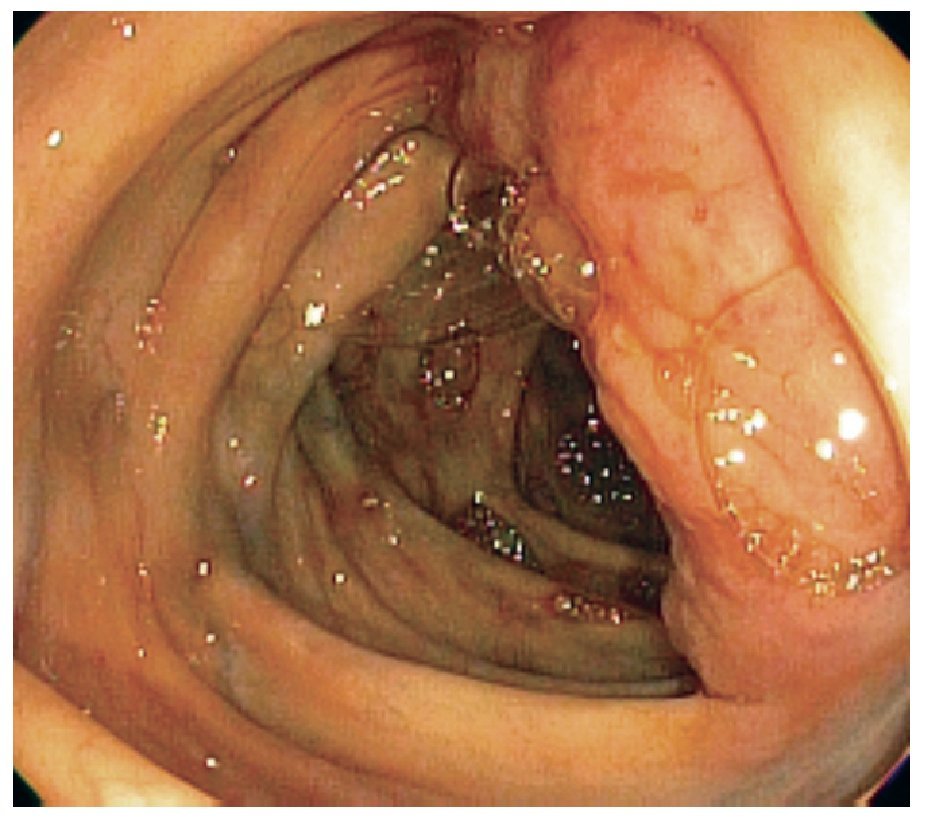
¡ð Figura 2.Colonoscopia, en la cual se demostró tumoración en colon ascendente, no obstructivo, de bordes definidos.
Se realizó laparotomía exploradora con exéresis de la masa tumoral, hemicolectomía de 15 cm de colon ascendente con anastomosis término-terminal. El tumor era de características: lobulado, de color rojizo, que al corte infiltraba la pared en forma transmural con infiltración a la válvula ileocecal e íleon terminal, perforado, con peritonitis aguda mucopurulenta, identificando ganglios peritumorales, por lo cual se procedió a resección de cinco ganglios afectados. Se realizó estudio de patología e inmunohistoquímica, los cuales mostraron como resultado linfoma de la zona marginal con diferenciación plasmocitoide inmunofenotipo B CD20 positivo, con restricción de cadenas ligeras lambda monoclonal (Figura 3). Se practicó serología de Helicobacter pylori, la cual dio resultados positivos. No se realizó endoscopia de tubo digestivo alto.
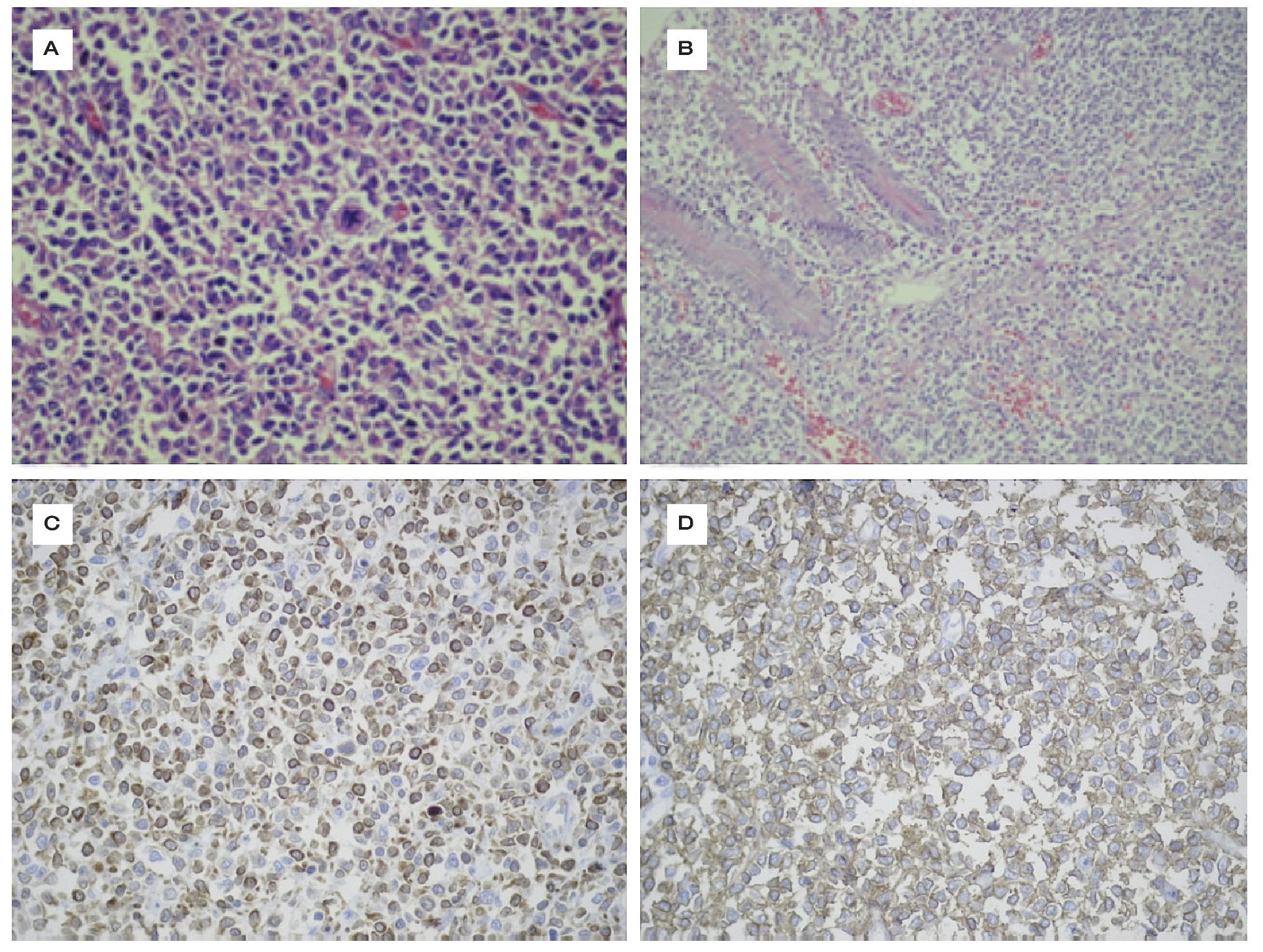
¡ð Figura 3. Linfoma de colon con mitosis A), Linfoma infiltrando mucosa B), Inmunohistoquímica CD 20+ C), Inmunohistoquímica con bcl2 positivo D).
Posterior a la cirugía, se practicó una tomografía por emisión de positrones, la cual mostró actividad intensa en retroperitoneo. La paciente fue tratada posteriormente con tres ciclos de quimioterapia tipo CHOP (ciclofosfamida, doxorubicina, vincristina y prednisona). Actualmente continúa en seguimiento a 13 meses del diagnóstico, demostrando ausencia de actividad tumoral.
Descripción y hallazgos del procedimiento
Se realizó laparotomía, en la que se encontró macroscópicamente un tumor pélvico benigno y una tumoración en colon ascendente, que ocupa el 50% de la superficie interna del intestino, el cual medía 5 x 4 x 3 cm, lobulado, de color rojizo. Al corte, el tumor infiltra la pared en forma transmural llegando a la válvula ileocecal en íleon terminal, identificando ganglios peritumorales. Se tomaron muestras para patología e inmunohistoquímica, la cual mostró como resultado linfoma de la zona marginal con diferenciación plasmocitoide inmunofenotipo B CD20 positivo con restricción de cadenas ligeras lambda monoclonal, perforado con peritonitis aguda mucopurulenta (Figura 3).
Discusión
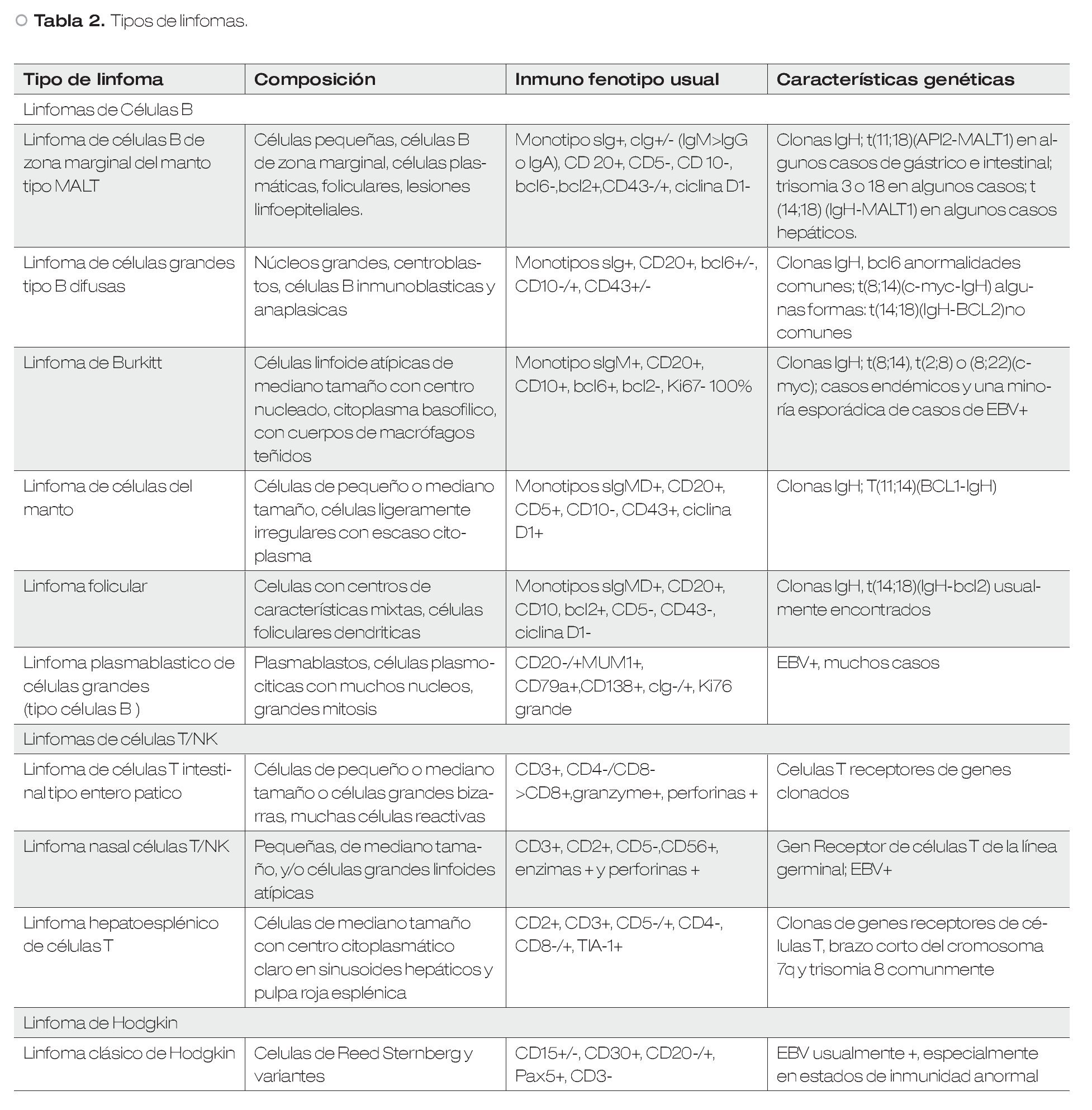
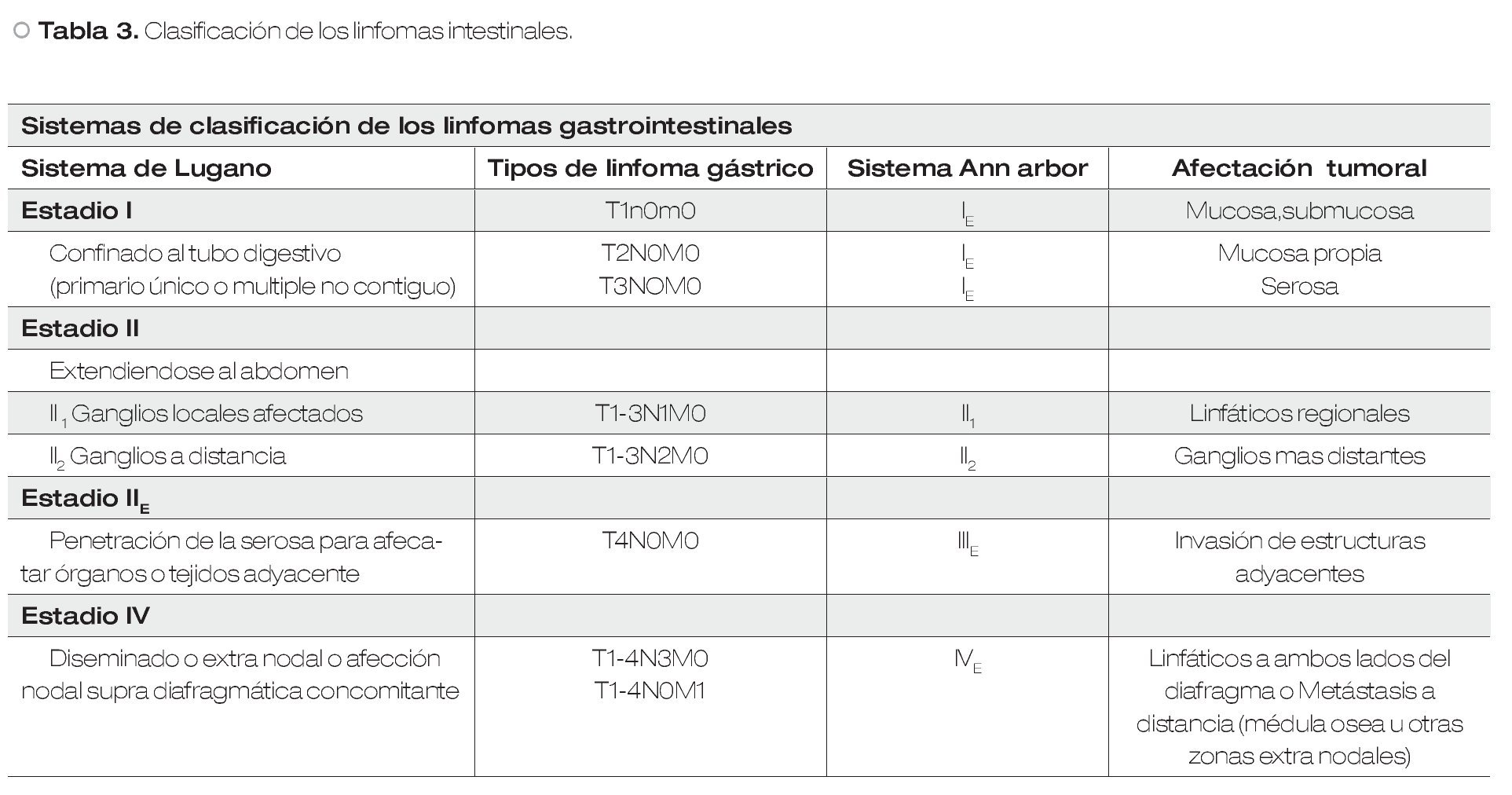
Dado que los linfomas ganglionares pueden afectar al tracto digestivo, se han establecido criterios diagnósticos (Criterios de Dawson, Tabla 1) para definir el linfoma primario de tracto gastrointestinal. Se han establecido clasificaciones, como la que se presenta en la Tabla 2,4 para estadificar los principales tumores a nivel de aparato digestivo de tipo linfoide (Tabla 3).

El linfoma de bajo grado de células B no Hodgkin extraganglionar surge de la MALT. Siendo una entidad recientemente reconocida, con características histológicas, citogenéticas e inmunohistoquímicas particulares. Las células tumorales encontradas de forma típica son: CD20+, CD79a+, CD5-, CD10-, CD23-, CD43+/-, CD11c+/-, CD21+ y CD35+.5 Citogenéticamente existen tres translocaciones típicas, que se observan en cerca del 35% de todos los linfomas tipo MALT, estos son: t(11;18) (q21;q21). t(1,14) (p22,q32), t(14;18) (q32;q21).6
Los linfocitos se originan en la médula ósea y están programados para llevar a cabo funciones específicas, las células T y B se desarrollan dentro de los tejidos linfoides primarios (timo de las células T y la médula ósea para las células B), y luego se diferencian en tejidos linfoides secundarios (ganglios linfáticos, el bazo, las amígdalas y tejido MALT).7
El tejido linfoide se encuentra distribuido en las superficies mucosas en forma de parches no encapsulados, lo que constituye el MALT. En el intestino puede ser un componente normal de distribución multifocal, teniendo como función proteger la mucosa intestinal de la penetración de antígenos.8
Los linfomas tipo MALT surgen de la zona marginal de los folículos linfoides, se caracterizan por la presencia de un infiltrado de linfocitos pequeños y una zona periférica rica en células. Se puede producir invasión local y destruir el epitelio glandular, con la formación de lesiones linfoepiteliales características.
Al producir invasión local, las células centrocíticas destruyen el epitelio glandular y forman lesiones linfoepiteliales, que son características de los linfomas tipo MALT.9
Es posible que el estado inflamatorio causado por la artritis reumatoide y el uso crónico de inmunosupresores en nuestro caso, haya contribuido a la producción de este tumor. Se ha observado que enfermedades como el síndrome de Sjögren, lupus eritematoso y la tiroiditis de Hashimoto, pueden predisponer a la aparición de linfomas tipo MALT. Estos pueden presentarse también en las glándulas salivales, tiroides y en el aparato respiratorio, así como en el timo, mama, próstata y riñón.10 Los estados inflamatorios crónicos y enfermedades autoinmunes, pueden actuar como estimuladores antigénicos de la mucosa del tracto digestivo,11 aunque el mecanismo exacto y la patogénesis involucrados en el desarrollo de este tumor no se conoce. Para que el linfoma se trasforme y progrese, se requiere la aparición de mutaciones genéticas. La evidencia reciente sugiere que sujetos con linfomas tipo MALT y con H. pylori negativos, pueden sufrir regresión con la terapia de erradicación de H. Pylori convencional, aunque esto no está claramente demostrado cuando ocurre en órganos extra gástricos. El manejo de estos casos incluye quimioterapia, radioterapia y resección quirúrgica.
Se ha informado que sólo el 2.5% de todos los linfomas tipo MALT de aparato digestivo, aparecen en el colon.12 En la mayoría de estos casos, se presentan como una sola masa y la aparición de la lesión es en general saliente y ulcerada, a diferencia de lo ocurrido en el caso que informamos, en quien el examen endoscópico reveló una lesión no ulcerada, de bordes bien definidos.
El estudio endoscópico fue de vital relevancia en este caso, para toma de biopsias de forma primaria antes de decidir la realización de extirpación quirúrgica del tumor.
Correspondencia: Dra. Ariana Paola Canché.
Hospital Ángeles del Pedregal, Camino a Santa Teresa 1055, Col. Héroes de Padierna. C.P. 10700. México D.F., México.
Teléfono: (55) 5449 5500, Fax: 5568 7847.
Correo electrónico: ariana_canche@hotmail.com