










El virus de la inmunodeficiencia humana tipo 1 (VIH-1) es el agente productor del sida una enfermedad reconocida desde hace 30 años que ha alcanzado proporciones pandémicas. Su origen se remonta a la transmisión a humanos de retrovirus que infectan a poblaciones de chimpancés en África central hace aproximadamente 100 años. Desde esta localización su expansión a todo el mundo ha sido espectacular principalmente en las últimas décadas. La intensa investigación realizada nos permite disponer de un tratamiento eficaz para controlar la replicación del virus y evitar la progresión de la enfermedad sin embargo no disponemos aún de una vacuna que impida la continua extensión de la pandemia. No es posible entender estos fenómenos sin un conocimiento detallado de la biología del VIH-1 y los mecanismos que se han seleccionado en este asombroso agente para infectar una célula clave como el linfocito T CD4+ y evadir la respuesta inmune.
The human immunodeficiency virus type 1 (HIV-1) is the agent that causes AIDS, a disease known for 30 years that has reached pandemic proportions. Its origin dates back to human transmission of retroviruses infecting populations of chimpanzees in central Africa about 100 years ago. From this location its expansion to the whole world has been phenomenal, particularly in recent decades. Extensive research has led to an effective treatment for controlling virus replication and to prevent progression of the disease, but we do not yet have a vaccine to prevent the continuing spread of the pandemic. It is not possible to understand these phenomena without detailed knowledge of the biology of HIV-1 and the mechanisms that have been selected in this amazing agent to infect a key cell such as the CD4 + T cell and evade the immune response.
El primer miembro de los Retrovirus fue inicialmente descrito en 1911 por Rous como un agente filtrable, más pequeño que una bacteria, capaz de transmitir la producción de tumores en pollos: el virus del Sarcoma de Rous. Posteriormente en 1970 Howard Temin y David Baltimore1,2 realizaron independientemente el descubrimiento central del mecanismo de retrotranscripción. La caracterización de esta nueva enzima, la retrotranscriptasa (RT) que permitía sintetizar ADN a partir de ARN, cuestionaba el dogma prevalente hasta el momento en la biología molecular que establecía que la expresión del gen siempre se realizaba en el sentido ADN→ARN→Proteína. La RT explicaba por qué esta creciente familia de virus ARN podía convertir su genoma en ADN e integrarlo como un gen más en el cromosoma de la célula infectada. Ahora sabemos que este mecanismo ha sido compartido a lo largo de la evolución por diferentes retrovirus y otros retroelementos de tal forma que en la actualidad más del 10% del genoma humano tiene este origen. En los años posteriores se describieron numerosos agentes retrovirales relacionados en su mayoría con tumores en aves y ratones. En 1980 Robert Gallo y su grupo, en plena expansión de las teorías del origen vírico de los tumores, descubren el primer retrovirus humano, el HTLV-I, un agente relacionado inicialmente con leucemia de células T y posteriormente con un cuadro neurológico conocido como paraparesia espástica tropical. Este mismo grupo describiría al año siguiente otro agente, el HTLV-II, relacionado esta vez con una rara leucemia de células peludas. Este acontecimeinto es doblemente relevante en la historia del VIH y el sida, porque si bien la investigación sobre los retrovirus humanos eataba ya plenamente establecida, precisamente el hecho de que se trataran en su mayoría de virus oncogénicos fue motivo de confusión inicial en la interpretación de la patogenia de la infección por VIH, un retrovirus cuya caracteristica principal es la destrucción del linfocito T CD4+, y no la transformación e inmortalización celular. El VIH se descubrió en 1983, a los dos años de la comunicación de los primeros casos de sida, por el grupo de Françoise Barré-Sinoussi y Luc Montagnier en el Insituto Pasteur de París3 y posteriormente en 1984 por el propio grupo de Robert Gallo en el Instituto Nacional de Cancer en Bethesda, EE.UU.4. Después de una importante polémica sobre la autoría del descubrimento, y la diferente nomenclatura propuesta por cada unos de los laboratorios, en 1986 se acordó la denominación de virus de la inmunodeficiencia humana.
Origen del virus de la inmunodeficiencia humanaEl VIH pertenece a la familia de los lentivirus y se clasifica en dos tipos: VIH-1 y VIH-2 que tienen un 40-50% de homología genética y una organización genómica similar (fig. 1). El VIH-1 es el causante de la pandemia mundial de sida mientras que el VIH-2, aunque también puede producir sida, se considera menos patogénico y menos transmisible. El VIH-2 se encuentra confinado principalmente a zonas del África occidental, aunque se han detectado algunos casos en Europa y EE.UU. Tanto el VIH-1 como el VIH-2 provienen de diferentes saltos inter-especie de virus que infectan en la naturaleza a poblaciones de simios en África. El VIH-2 está muy cercano filogenéticamente al SIVsm, virus de la inmunodeficiencia del Sooty mangabey, una variedad de mono muy frecuente en Africa occidental. El origen del VIH-1ha sido mucho más laborioso de esclarecer ya que proviene del agente que infecta en la naturaleza a la variedad de chimpancé Pan troglodytes troglodytes que habita en zonas poco accesibles del sur de Camerún (SIVcpzPtt)5,6. Las cepas del VIH-1 se han clasificado en tres grandes grupos según su homología genética y se piensa que representan diferentes episodios de salto inter-especies. Estos son el grupo M (main o principal), el grupo O (outlier), y el grupo N (no M, no O). El grupo M se ha dividido en 9 subtipos (A, B, C, D, F, G, H, J, K) y en cepas recombinantes entre ellos, denominados CRF (formas recombinantes circulantes). Los CRF se forman por recombinación de fragmentos genómicos de distintos subtipos. Actualmente se han descrito más de 30 CRF y su número se incrementa constantemente.
. El que infecta al Sooty mangabey (SIVsm) es el origen del VIH-2 en humanos. El VIH-1 grupo M procede del virus (SIVcpz-ptt) que infecta a una de las 4 variedades de chimpancé (Pan troglodytes troglodytes) que habita en bosques del sur de Camerún. El ancestro del VIH-1 grupo N también se ha encontrado en chimpancés de esa misma zona. El origen del grupo O está menos claro, ya que hasta el momento no se ha relacionado con ningún aislamiento en chimpancé y parece más relacionado con el SIVgor que infecta a poblaciones de gorila. Recientemente se ha identificado la infección en humanos por un VIH-1 más cercano que el grupo O al SIVgor y se ha propuesto la denominación de grupo P.")
En África se han descrito más de 30 especies de monos infectados naturalmente con variedades de virus de la inmunodeficiencia del simio (SIV). El que infecta al Sooty mangabey (SIVsm) es el origen del VIH-2 en humanos. El VIH-1 grupo M procede del virus (SIVcpz-ptt) que infecta a una de las 4 variedades de chimpancé (Pan troglodytes troglodytes) que habita en bosques del sur de Camerún. El ancestro del VIH-1 grupo N también se ha encontrado en chimpancés de esa misma zona. El origen del grupo O está menos claro, ya que hasta el momento no se ha relacionado con ningún aislamiento en chimpancé y parece más relacionado con el SIVgor que infecta a poblaciones de gorila. Recientemente se ha identificado la infección en humanos por un VIH-1 más cercano que el grupo O al SIVgor y se ha propuesto la denominación de grupo P.
Por medio del estudio evolutivo de secuencias se piensa que el SIVcpz pasó del chimpancé a la especie humana alrededor de 1900. El mecanismo de exposición más probable ha sido la caza y el consumo de carne de chimpancé, práctica muy popular en la zona donde se han descrito infecciones en humanos de agentes que son característicos de simios como SFV (Espumavirus de simio) y nuevas variedades de HTLV7; estos virus no tienen potencial patogénico aparente pero son marcadores de la transmisión de agentes entre simios y humanos. La infección en humanos por el VIH-1 probablemente se mantuvo inicialmente limitada a pequeños grupos de población hasta que alcanzó, seguramente a través del Río Congo, un núcleo urbano en rápida expansión como era la ciudad de Kinshasa alrededor de 1930-408. En esta ciudad existe la mayor variedad de cepas y los indicios de la divergencia del virus en una nueva especie, los humanos, en lo que hoy conocemos como subtipos. A partir de este punto el VIH se diseminó por el continente por contacto sexual, y muy probablemente por prácticas sanitarias con material contaminado, hasta que se introdujo en el mundo desarrollado durante los años setenta, causando los primeros casos de sida detectados inicialmente en EE.UU. a principios de los ochenta. El VIH-1 grupo M es el reponsable principal de la pandemia de sida. Dentro de este grupo, las cepas del subtipo B predominan en Europa y América y son poco frecuentes en África. Este hecho es todavía motivo de discusión aunque la explicación más probable es que el VIH-1 subtipo B entrase en EE.UU, y posteriormente en los países desarrollados, vía Haití. De nuevo el estudio evolutivo de las secuencias nos da la clave: los aislamientos de VIH-1 en Haití durante los años ochenta son las secuencias ancestrales del subtipo B y han dado lugar a los primeros aislamientos en EE.UU. y posterioremente en Europa, Australia y Japón. En el Congo pos-colonial francófono está confirmada la presencia de nativos de Haití en tareas de cooperación sanitaria y educación. La hipótesis más probable es que durante los años sesenta unos pocos individuos, incluso un solo individuo, de Haití llevase una variedad muy particular de VIH-1, el subtipo B, desde Congo hasta Haití donde se expandió e introdujo en EE.UU. a finales de los años sesenta, dando lugar a una rapidísima diseminación9. Actualmente, y en relación principalmente con la inmigraciónal, al menos el 25% de las nuevas infecciones en Europa se producen por variantes no-B procedentes de África y Asia, siendo los subtipos A, C y los recombinantes CRF01_AE y CRF02_AG las variantes más frecuentes (fig. 2). Los virus N y O, han pasado también a la especie humana pero no han tenido diseminación epidémica y han dado solo lugar a unos pocos casos de infección en humanos detectados principalmente en África occidental. Recientemente se ha descrito un cuarto grupo de VIH-1, denominado “P”. Este virus está más cercano filogenéticamente al SIVgor que infecta al gorila occidental (Gorilla gorilla) habitante de las mismas áreas donde se han identificado chimpancés infectados con los ancestros del grupo M y N10. Las vías de la trasmisión entre chimpaces y gorilas del ancestro del grupo P, y posiblemente del O, no están totalmente aclaradas.
 y VIH2.")
En España, al igual que en el resto de Europa y América, predomina el subtipo B, aunque se está observando una circulación creciente de subtipos no-B en los últimos años, principalmente los subtipos G y el CRF02_AG, encontrados sobre todo en pacientes procedentes de África occidental aunque, como en la mayoría de los paises esuropeos, se han descrito casos de todos los subtipos en población no inmigrante (fig. 2).
Características estructurales del VIHLa envoltura del VIHEl VIH-1 tiene forma de esfera con un diámetro de 100-120nm. Al igual que en todos los virus envueltos, la envoltura consiste en una bicapa lipídica tomada de la membrana de la célula humana durante el proceso de gemación de nuevas partículas. En esta envoltura se encuentran presentes algunas proteínas de la célula huésped y muy significativamente Env, la glicoproteina de envoltura del VIH. Env se encuentra anclada en la membrana y consiste en un hetero-trímero formado por tres moléculas llamadas glicoproteína 120 (gp120), en la zona más externa, y un tronco de una estructura transmembrana que consta a su vez de tres moléculas llamadas glucoproteína 41 (gp41) (fig. 3). La estructura y funcionalidad de Env son claves para entender aspectos importantes de la biología del VIH-1, tales como la interacción con receptores celulares (tropismo) y la evasión inmune. Cada partícula de VIH-1 tiene una cantidad de estas estructuras de Env (spikes) relativamente pequeña: 14±711, a lo que se añade su fragilidad al ser la unión entre gp120 y gp41 no-covalente. Este factor probablemente es responsable de la fragilidad y corta infectividad de las partículas de VIH-1 ya que la mayoría de los spikes no son funcionales. No obstante, el diseño de la envuelta encierra algunas ventajas biológicas muy especiales de tal forma que precisamente Env es en gran parte responsable de que todavía no exista una vacuna protectora frente a la infección por VIH-1. Los factores relacionados con la dificultad de neutralizar la infección por VIH-1 están directamente relacionados con Env: 1) gran variabilidad de la envoltura con 5 regiones hipervariables en la zona más externa de gp120, (2) alto nivel de glicosilación de Env con más del 50% de su masa en azúcares (N-glicosilación), que impide la unión de anticuerpos (escudo de glicanos)12, y 3) enmascaramiento conformacional13, término que describe el que una de las zonas más vulnerables de Env, el sitio de unión con los co-receptores (CCR5 ó CXCR4), no existe hasta que se organiza espacialmente después del cambio en la conformación de gp120 inducido por la interacción con CD4, y es por tanto muy poco susceptible a la neutralización mediada por anticuerpos (fig. 4).
 que puede recubrir una partícula de 100-120nm es de más de 70, las estimaciones más recientes indican que este número es mucho más bajo con una media de 14 spikes por virión.")
Esquema general de la estructura de la partícula de VIH-1. ENV: Envoltura, MA: Matriz, CA: Cápside, NC: Nucleocápside, IN: Integrasa, RT: Retrotranscriptasa, PR, Proteasa, ARN: Genoma del virusAunque el número de spikes de Env (gp120/gp41) que puede recubrir una partícula de 100-120nm es de más de 70, las estimaciones más recientes indican que este número es mucho más bajo con una media de 14 spikes por virión.
. Gp120 interacciona con las moléculas CD4 y CCR5 o CXCR4 (B) y se produce un cambio conformacional secuencial que activa los dominios fusogénicos de gp41 que median la fusión entre las membranas del virus y la célula (D). La subunidad gp120 presenta en el exterior zonas hipervariables y una abundante glicosilación que dificulta la neutralización por anticuerpos. La zona responsable del reconocimiento de CD4 es poco accesible así como la zona de unión al co-receptor CCR5 o CXCR4 que solo se constituye espacialmente después de la interacción con CD4 (B).")
La estructura principal de la envoltura del VIH consiste en un trímero de gp120 y gp41 anclado en la membrana externa (C). Gp120 interacciona con las moléculas CD4 y CCR5 o CXCR4 (B) y se produce un cambio conformacional secuencial que activa los dominios fusogénicos de gp41 que median la fusión entre las membranas del virus y la célula (D). La subunidad gp120 presenta en el exterior zonas hipervariables y una abundante glicosilación que dificulta la neutralización por anticuerpos. La zona responsable del reconocimiento de CD4 es poco accesible así como la zona de unión al co-receptor CCR5 o CXCR4 que solo se constituye espacialmente después de la interacción con CD4 (B).
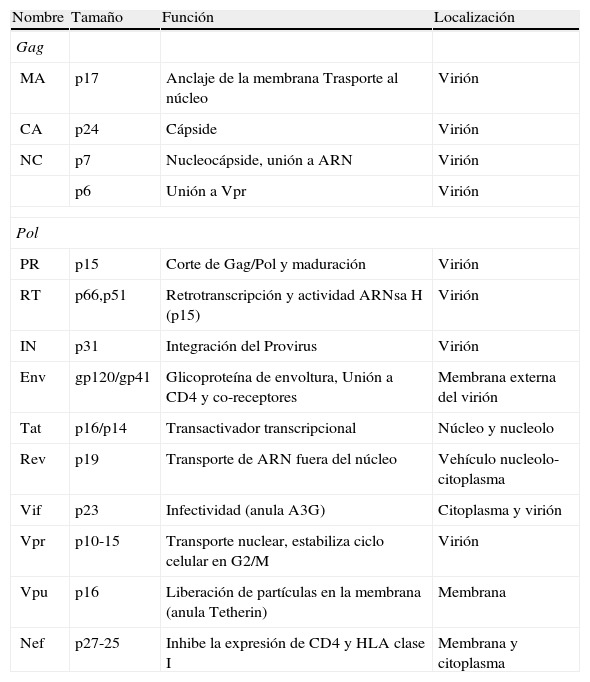
El gen gag codifica las principales proteínas estructurales: la proteína de matriz p17, anclada en el interior de la membrana y la proteína de la cápside p24, que forma por polimerización una estructura nuclear cónica que contiene en su interior un complejo proteína-ácido nucleico formado por dos copias del ARN genómico del VIH-1, la nucleoproteína p7 y la transcriptasa inversa p66 (RT). El gen pol codifica los tres enzimas necesarios para el ciclo infectivo del virus: la proteasa (PR), la transcriptasa inversa (RT) y la integrasa (IN) (fig. 5). Además, el VIH-1 contiene otros seis genes denominados inicialmente accesorios: tat, rev, nef, vif, vpu y vpr, que dan lugar a sus correspondientes proteínas con un papel muy importante en el ciclo biológico del virus (tabla 1).
 compuestas or las regiones U3, R y U5. El LTR 5¿controla la expresión de los genes estructurales: gag, pol y env, y los accesorios: tat, rev, nef, vif, vpu y vpr. MA: Matriz, CA: Cápside, NC: Nucleocápside, IN: Integrasa, RT: Retrotranscriptasa, PR: Proteasa. Los 9 genes de VIH-1 se expresan por medio de diferentes mensajeros a partir del provirus (ADN) integrado. Los ARN más largos son exportados al citoplasma por un mecanismo Rev-dependiente y constituyen el genoma de nuevas partículas o son traducidos a las proteínas estructurales Gag, Pol y Env. Las proteínas reguladoras o accesorias son producto de extenso procesamiento (splicing) del ARN en el núcleo donde se producen hasta 7 versiones diferentes del ARN transcrito.")
Organización del genoma de VIH-1. En la forma proviral el virus integrado esta flanqueado por las regiones terminales repetidas (LTR) compuestas or las regiones U3, R y U5. El LTR 5¿controla la expresión de los genes estructurales: gag, pol y env, y los accesorios: tat, rev, nef, vif, vpu y vpr. MA: Matriz, CA: Cápside, NC: Nucleocápside, IN: Integrasa, RT: Retrotranscriptasa, PR: Proteasa. Los 9 genes de VIH-1 se expresan por medio de diferentes mensajeros a partir del provirus (ADN) integrado. Los ARN más largos son exportados al citoplasma por un mecanismo Rev-dependiente y constituyen el genoma de nuevas partículas o son traducidos a las proteínas estructurales Gag, Pol y Env. Las proteínas reguladoras o accesorias son producto de extenso procesamiento (splicing) del ARN en el núcleo donde se producen hasta 7 versiones diferentes del ARN transcrito.
Tamaño, localización y función de las proteínas que codifican los 9 genes de VIH-1.
| Nombre | Tamaño | Función | Localización |
| Gag | |||
| MA | p17 | Anclaje de la membrana Trasporte al núcleo | Virión |
| CA | p24 | Cápside | Virión |
| NC | p7 | Nucleocápside, unión a ARN | Virión |
| p6 | Unión a Vpr | Virión | |
| Pol | |||
| PR | p15 | Corte de Gag/Pol y maduración | Virión |
| RT | p66,p51 | Retrotranscripción y actividad ARNsa H (p15) | Virión |
| IN | p31 | Integración del Provirus | Virión |
| Env | gp120/gp41 | Glicoproteína de envoltura, Unión a CD4 y co-receptores | Membrana externa del virión |
| Tat | p16/p14 | Transactivador transcripcional | Núcleo y nucleolo |
| Rev | p19 | Transporte de ARN fuera del núcleo | Vehículo nucleolo-citoplasma |
| Vif | p23 | Infectividad (anula A3G) | Citoplasma y virión |
| Vpr | p10-15 | Transporte nuclear, estabiliza ciclo celular en G2/M | Virión |
| Vpu | p16 | Liberación de partículas en la membrana (anula Tetherin) | Membrana |
| Nef | p27-25 | Inhibe la expresión de CD4 y HLA clase I | Membrana y citoplasma |
CA: cápside; IN: integrasa; MA: matriz; NC: nucelocápside; PR: proteasa; RT: retrotranscriptasa.
Tat y Rev son proteínas reguladoras que se acumulan en el núcleo y se unen a regiones específicas del ARN viral: TAR y RRE respectivamente. La proteína Tat es un potente activador de la transcripción y es esencial para la replicación del virus. Rev es un factor de exportación nuclear que facilita la salida al citoplasma de los ARN mensajeros largos antes de ser procesados en el núcleo y permite así la traducción y expresión de las proteínas estructurales.
La proteína Nef ha sido motivo de numerosas investigaciones ya que participa en funciones diversas. Nef induce regulación negativa de CD4 y moléculas HLA de clase I en la superficie de las células infectadas14, lo que puede representar un mecanismo de escape importante al evadir un ataque mediado por linfocitos CD8 + citotóxicos. Nef también parece interferir con la activación del linfocito T al unirse a varias proteínas que intervienen en las vías de transducción de señales intracelulares. Vpr es importante para el transporte al núcleo del complejo viral pre-integración, inmediatamiente después de la entrada del virus a la célula lo que permite al VIH-1, a diferencia de la mayoría de los retrovirus, infectar células que no estén activamente dividiéndose.
y algunos antídotos: El papel principal de Vif ha quedado claro que consiste en interaccionar con una proteína de defensa antiviral en la célula humana llamada APOBEC3G (A3G)15. Vif interfiere con la actividad de A3G, anulando su efecto antiviral y favoreciendo la replicación del VIH. A3G era una proteína conocida pero su función no estaba clara. Este tipo de moléculas celulares con efecto antiviral se conoce como “factores de restricción” y parecen constituir un nuevo sistema de defensa intracelular frente a diferentes agentes virales. A3G es un deoxi-citidina deaminasa que actúa sobre el ADN recién sintetizado por retro-transcripción y modifica así las citosinas en uracilos. Estos cambios C por U tiene como resultado bien la destrucción de la cadena por el enzima UNG (solo se permite U en cadenas de ARN) o si la cadena escapa a la destrucción se produciría el cambio paulatino de residuos G por A (hipermutación) con la consiguiente acumulación de mutaciones. El estudio de esta interacción puede servir como una nueva diana para los fármacos antivirales. Recientemente se ha descrito que Vpu, otra de estas llamadas proteínas accesorias, tiene, al igual que Vif, la función de interferir con un nuevo factor de restricción denominado Tetherin y que tiene como mecanismo antiviral la misión de impedir la liberación de partículas de la membrana celular16.
Entrada, tropismo y ciclo infectivoDesde el la descripción de los primeros casos de sida en EE.UU., y muy rápidamente en Europa y resto del mundo, fue muy llamativa la intensa depleción de linfocitos T CD4+ que presentaban los pacientes. Es precisamente esta la célula diana principal del virus y expresa en la superficie los dos receptores necesarios para la entrada: la propia molécula CD4 y un receptor de quimiocinas, generalmente CCR5 en las primeras fases de la infección. En algunos pacientes el virus puede utilizar un receptor alternativo CXCR4 en fases avanzadas de su evolución, en lo que se conoce como cambio de tropismo. La molécula gp120 experimenta un cambio conformacional al interaccionar con CD4 y se produce entonces una segunda interacción con el receptor de quimiocinas CCR5 o CXCR4. Este doble reconocimiento de receptores induce la exposición de la zona fusogénica amino-terminal de gp41, el otro componente de la envoltura, que permite la fusión de las membranas viral con la celular y la entrada de la partícula.
Aunque CD4 fue rápidamente identificado en 1984 como un receptor necesario para la entrada de VIH en la célula humana17 y existía la evidencia de que se necesitaban moléculas alternativas para completar la infección, la caracterización de los co-receptores CXCR4 y CCR5 se prolongó 12 años más a pesar del esfuerzo de un buen número de laboratorios18,19. CCR5 es el co-receptor utilizado por las cepas de VIH que se transmiten e inician la infección en la práctica totalidad de los casos20–22. Estas cepas con tropismo por CCR5 (R5 trópicas) se mantienen detectables a lo largo de la evolución de aproximadamente la mitad de los pacientes infectados22. En el resto, y coincidiendo con fases avanzadas de la enfermedad, se detectan variantes que utilizan el co-receptor CXCR4 (X4 trópicas), existiendo también cepas duales que pueden utilizar los dos receptores23. Este patrón temporal en el tropismo de VIH por los dos co-receptores sigue siendo un enigma y desconocemos en su mayor parte por qué la infección comienza invariablemente por cepas R5 y cuáles son los factores determinantes del cambio de tropismo en gran parte de los pacientes infectados. En cambio las bases moleculares que determinan el tropismo se conocen con bastante detalle. La utilización de CCR5 o CXCR4 depende fundamentalmente de la secuencia de la tercera región variable de la envuelta, V3, y dentro de esta de los aminoácidos en las posiciones 11 y 2524,25 donde la incorporación de aminoácidos básicos, como R o H, determinaría el tropismo X4. Recientemente se han identificado cambios fuera de estas regiones, en V2 e incluso en gp41, que podrían jugar un papel menor en la utilización de los co-receptores26. El hecho de que un pequeño número de cambios determine el tropismo podría anticipar un rápido cambio hacia la utilización de una molécula mucho más abundante como es CXCR4. Esto contrasta con la evidencia de que el cambio de tropismo se produce solo en fases avanzadas y no en todos los pacientes. La sustitución de cepas R5 por cepas X4 está claramente relacionada con progresión de la enfermedad y el debate continúa sobre si la emergencia de cepas que utilizan CXCR4 es causa o más bien consecuencia o de la inmunodeficiencia27. El tropismo parece no afectar al tratamiento aunque la existencia de cepas X4 se relacionan significativamente con menores cifras de CD4 y eventos clínicos relacionados con sida27 Parece claro que biológicamente las cepas X4 tienen alguna limitación selectiva para transmitirse y replicarse en el contexto de un sistema inmunológico competente28. Algo de luz sobre este fenómeno puede venir de la virología comparada. Los lentivirus de primates parecen tener exclusivamente tropismo R529 y de hecho la aparición de cepas X4 parece ser más frecuentes en VIH subtipos B y D que en otros subtipos30,31. Se han caracterizado algunas cepas con muy baja afinidad por CD4 e incluso CD4 independientes pero que siguen reconociendo CCR532, estas cepas CD4 independientes parecen ser mas vulnerables a la neutralización por anticuerpos al igual que ocurre en las cepas X4, lo que podría explicar en parte su ausencia en infección primaria29,33. Todo esto induce a pensar que CCR5 es el receptor principal de lentivirus de primates, incluyendo VIH, y ayudaría a entender por qué el cambio de tropismo hacia X4 no es un fenómeno generalizado y no constituye el mecanismo principal de desarrollo de resistencias a antagonistas de CCR5. Una interesante observación, que apoya la idea de que las cepas con tropismo X4 tienen alguna desventaja selectiva en relación a las R5 se ha realizado en algunos pacientes en tratamiento con MVC. Estos pacientes que presentaban inicialmente cepas mayoritariamente R5 pero durante el tratamiento seleccionaban variantes minoritarias X4 pre-existentes, al suspender la administración de MVC las cepas R5 de nuevo recuperaban su presencia mayoritaría34.
La primera partículaEl VIH-1 se transmite principalmente por contacto sexual a través de la gran concentración de partículas en semen y fluidos genitales. Existe un gran interés en aclarar los mecanismos que determinan la transmisión del virus en la superficie de las mucosas genitales. Sin embargo este acontecimiento ha sido muy difícil de estudiar ya que la infección es prácticamente imposible de detectar hasta por lo menos 1-2 semanas después de la primoinfección. A través del seguimiento y la obtención de muestras periódicas de cohortes de parejas discordantes principalmente en África, se ha podido disponer de muestras clínicas de infecciones en momentos muy recientes. Algunos grupos han estudiado estas muestras mediante técnicas de secuenciación clonal, es decir secuenciando por dilución límite genomas de partículas individuales de VIH-1. Por medio del estudio de secuenciación clonal se puede establecer de manera mucho más precisa la evolución de las secuencias circulantes y, si se dispone de muestras en fases muy tempranas, es posible reconstruir la evolución de las secuencias retrospectivamente y llegar a identificar las secuencias fundadoras que representarían las partículas que se han transmitido e iniciado la infección. Varios trabajos principalmente del grupo de George Shaw en la Universidad de Alabama en Birmingham35,36 han confirmado que la primoinfección se produce a expensas de una sola partícula (secuencia) en más del 80% de los casos estudiados, siendo el resto producidos por un número muy reducido de secuencias. El origen monoclonal u oligoclonal de la infección por VIH-1 es coherente con el riesgo estimado de transmisión por contacto heterosexual que se ha estimado en 1/200-300 y ha estimulado el interés por encontrar las propiedades especiales de esas partículas para la transmisión. Hasta el momento solo se ha encontrado el tropismo R5 como característica uniforme. Utilizando el mismo abordaje experimental se ha encontrado recientemente una mayor frecuencia de transmisones múltiples en varones homosexuales recientemente infectados37 (36% con más de una cepa fundadora versus 19% en heterosexuales) lo que de nuevo podría ser compatible con un mayor riesgo de infección en relación con el sexo anal y un mayor reto para la protección de una vacuna.
Replicación y variabilidadUna de las caracteristicas de la replicación de los retrovirus y en particular del VIH-1 es su gran capacidad de variabilidad. El proceso de retrotranscripción tiene una relativa alta tasa de error (1 de cada 104 nucleótidos) a lo que se añade la facilidad para la recombinación de fragmentos genómicos si varías partículas infectan la misma célula. Si consideramos que en un paciente infectado se producen 1010-1012 partículas diarías, las posibilidades de que ocurra un cambio en una posición determinada son muy altas38,39. El VIH-1 se caracteriza por una elevada heterogeneidad genética lo que favorece que en la población de virus de un mismo individuo existan genomas relacionados entre sí, pero no idénticos y que se conocen como cuasiespecies víricas. Evolutivamente, la existencia de cuasiespecies es la consecuencia del proceso continuo de mutación, competición y selección de los genomas mejor adaptados a las condiciones que rodean al virus como puede ser la presión selectiva ejercida por el sistema inmune o la presencia de fármacos40,41. En teoría, todas estas variantes circulantes pueden integrarse en forma de provirus en las células y estar representadas en el reservorio de células latentemente infectadas. Si alguno de estos cambios confiere una ventaja selectiva como por ejemplo evasión de respuesta inmune o resistencia a los anti-retrovirales, esta secuencia tendría selección positiva. Este fenómeno de hecho ocurre constántemente y es una de las mayores dificultades con las que se enfrenta nuestro sistema inmunológico y el diseño de estrategias antivirales como vacunas o fármacos.
Latencia celular y viremia residualReservorios de VIHEl grupo de Robert Siliciano, Universidad Johns Hopkins, Baltimore, se ha distinguido por establecer la mayor parte de nuestro conocimiento del reservorio viral en la infección por VIH. El linfocito T CD4+ infectado se destruye en 24 horas al completar el virus un ciclo infeccioso, sin embargo unas pocas células infectadas no son destruidas y pueden revertir al estado quiescente después de la infección albergando al virus latente durante periodos muy prolongados, en lo que se conoce como reservorio. El reservorio celular latente de VIH-1 consiste principalmente en linfocitos T CD4+ memoria en estado de reposo. Utilizando técnicas muy precisas de PCR y cultivo se ha detectado VIH-1 integrado en 1/103-4 CD4+ memoria circulantes aunque en muchas de estas células el virus podría no ser viable ya que la proporción de reservorio con potencial de reactivación ex vivo se ha estimado en ≤1/106 de estas células42,43. Este reservorio de células latentemente infectadas tiene una vida media de más de 4 años y es el obstáculo principal para la erradicación del virus. La infección por VIH es intrínsecamente incurable con anti-retrovirales porque, aunque sea posible frenar completamente la replicación del virus durante largos periodos, al suspender el tratamiento se reinicia la replicación a expensas de este reservorio. Debido a las limitaciones técnicas para trabajar con cantidades tan pequeñas de células infectadas, prácticamente toda la información sobre latencia se refiere a sangre periférica y no podemos excluir la posibilidad de la existencia de algún reservorio latente en otras localizaciones no accesibles de momento a estudio experimental. Una de las posibilidades es que se trate de una célula con características de célula madre con vida larga y capacidad de auto-renovación44.
Viremia residualCaracterizar los mecanismos implicados en el mantenimiento del reservorio celular es un objetivo de la mayor importancia. Las tres hipótesis que se invocan no son completamente excluyentes: 1) Larga vida media de las células laténtemente infectadas, 2) Proliferación y expansión de algunas células del reservorio y 3) Persistencia de ciclos de replicación de muy bajo nivel. Mediante técnicas especiales capaces de detectar una sola copia de ARN de VIH-1ha sido posible comprobar que en la mayoría de los pacientes en tratamiento y con carga viral plasmática estándar indetectable (<50 copias/ml) existe una muy pequeña cantidad de partículas circulantes, generalmente 3-15 copias/ml, denominada viremia residual. Determinar el origen de esta viremia residual es clave, ya que si se tratase de partículas provenientes de ciclos de replicación no controlados sería esperable la evolución de secuencias y la emergencia de mutantes resistentes. La mayor parte de las evidencias experimentales indican más bien que está viremia residual es el producto de la liberación de partículas de forma estocástica por un pequeño número de células latentemente infectadas (fig. 6). La presencia de concentraciones efectivas de anti-retrovirales impediría que infectasen nuevas células susceptibles. Las dos evidencias principales que soportan está hipótesis son: 1) El análisis de las secuencias de la viremia residual a lo largo del tiempo no muestra un patrón de evolución temporal ni la selección positiva de nuevas mutaciones de resistencia y es más bien representativa de secuencias ancestrales presentes en el reservorio celular45,46 y 2) la intensificación del tratamiento anti-retroviral añadiendo nuevos compuestos no parece modificar el nivel de la viremia residual en la mayoría de los estudios realizados47–49. Es bastante razonable pensar que el tratamiento de combinación actual suprime completamente la replicación y la evolución del VIH y por tanto, en ausencia de toxicidad, los pacientes podrían mantenerse controlados virológicamente por tiempo indefinido.

Viremia residual. La viremia de VIH-1 superior a 50 copias/mL de plasma es el producto de ciclos replicativos de partículas que infectan células susceptibles. En pacientes en tratamiento supresor y con carga viral < 50 copias/ml es posible detectar pequeñas cantidades de partículas circulantes. Esta viremia residual estaría relacionada con liberación de partículas de células laténtemente infectadas y no completan nuevos ciclos infectivos.
Nota: sección acreditada por el SEAFORMEC. Consultar preguntas de cada artículo en: http://www.elsevier.es/eimc/formacion.

