




Introduction
Aspergillus fumigatus, a filamentous fungus which causes several pulmonary conditions such as allergic bronchopulmonary aspergillosis, aspergilloma and invasive aspergillosis, has emerged as a major problem in immunosupressed patients1. Nosocomial outbreaks of aspergillosis are being more frequent, and identification of the isolates may be delayed because of waiting for diagnostically appropiate structures. Furthermore, inexperience in microscopy may lead to misidentification.
Randomly amplified polymorphic DNA (RAPD) analysis assays DNA sequence variation in short regions using one short primer and a low annealing temperature to generate several fragments in one amplification reaction2. Moreover, RAPD analysis is technically simple and often detects variation among isolates that are invariant with Restriction Fragment Length Polymorphism (RFLP) analysis3,4. However, this method is gradually being abandoned because of poor reproducibility5. Recently, Ellinghaus et al. have communicated increased efficiency and reproducibility of arbitrarily primed PCR by prolonging ramp times6.
Otherwise, touchdown PCR with Z19-276 and Z19-660 primers described by Brandt et al.7 is a useful tool in molecular identification of Aspergillus fumigatus. This shortens the time, substantially, needed for fungal identification.
The present report describes the use of RAPD analysis as a reproducible genotyping method for A. fumigatus.
Methods
A total of 16 fungal strains isolated from the clinical and hospital environment were tested in this study. American Type Culture Collection (ATCC) 13073 A. fumigatus and ATCC 1004 A. niger were used as positive and negative control, respectively. Isolates were stored at 70 oC in brain heart infusion broth containing 10% glycerol. Working stocks were mantained on Sabouraud agar medium at 4 oC, and species identifications were made on the basis of standard morphological features8.
DNA extraction
DNA was extracted by a rapid small-scale method that allows several samples to be processed simultaneously. Briefly, a Sabouraud-dextrose agar in a petri dish was inoculated with conidia and incubated at 37oC during 24 to 48h. Mycelial growth was peeled off from the agar surface with a sterile scalpel and transferred to a 1.5 ml polypropylene screw-cap tube containing six glass beads. The tube was immersed in liquid nitrogen for 10 s and vortexed vigorously for 30s. Then ground fungal tissue was processed by E.Z.N.A. system (a purifying-column based method) (Omega Biotek, USA) according to the manufacturer´s instructions. The DNA concentrations were estimated spectrophotometrically.
Touchdown PCR
It was performed in a model 9600 DNA thermal cycler (Perkin-Elmer Corp., Applied Biosystems). PCR core reagents and polymerase were purchased from Perkin-Elmer. PCR reaction and amplification conditions were made as previously described7. Two primers were chosen to amplify the 864-bp A. fumigatus-specific fragment: primer Z19-276 (5'-TTGATCTGGCCCTGGCTTGGG) and primer Z19-660 (5'-CAACATTGAAATCCAAGAGGC). Amplification reactions were made in 50 µ L volumes containing 1 x PCR buffer (Perkin-Elmer), 2.5 mM MgCl2, 0.2 mM each deoxinucleotide triphosphate, 10 pmol each primer, 1 U AmpliTaq and 50 ng DNA. Amplification protocol was: 31 cycles of 1 minute at 94 ºC for denaturation, 1 minute at 65 ºC for the three begining cycles and, after, it was decreasing one grade each three cycles, and two grades from 59 ºC to 55 ºC, temperature used to perform the last 10 cycles. All cycles were finished with 1 minute at 72 ºC for elongation.
RAPD analysis
DNA typing by RAPD was assayed with two oligonucleotide primers, OPZ19 (5'-GTGCGAGCAA) and R108 (5'-GTATTGCCCT). Amplification reactions were made in 50 µ L volumes containing 50 mM KCl, 10 mM Tris-Cl (pH 8.0); 1.5 mM MgCl2; 100 µM each dATP, dCTP, dTTP, and dGTP (Perkin-Elmer), 0.2 µM each primer?; 50 ng of genomic DNA; and 2.5 U of Taq polymerase (Perkin-Elmer). Amplifications were carried out by two different protocols. The first one at 1 cycle of 5 minutes at 95 oC to denature followed by 45 cycles of 1 minute at 95 oC, 1 minute at 34 oC and 2 minutes at 72 oC (temperature/time rate as minimal as possible) and, finally, 10-minutes final extension at 72 oC. The second one was performed prolonging ramp times drastically between annealing and extension temperatures, 5 and 7 minutes, respectively. Amplification products were fractionated by electrophoresis through 1.8% agarose NuSieve 3:1 (FMC Bioproducts, USA) gels run in 0.5x Tris-borate-EDTA in the presence of ethidium bromide (0.5 mg/ml), photographed and analyzed. Lanes corresponding to the same sample in different runs were compared by superimposing their intensity profiles (the Rf values in X axis and the pixel intensity values in Y axis) using Quantity one vs. 4.0 software (Bio-Rad, USA). Afterwards, the similarity coefficient (S) was calculated according to S= 2 NAB/ NA + NB2
Results
Phenotypical identification of the fungal strains
A total of 16 fungal strains were isolated by culture techniques. Four A. fumigatus strains (strains number 1, 2, 3, 5) and 1 A. niger strain from clinical source. The following strains were recovered from the hospital environment: 5 A. fumigatus (strains number 7, 8, 9, 11, 13), 2 A. niger (strains number 14, 16), 1 Emericella quadrilineata (strains number 10), 1 Emericella nidulans (strain number 15), 1 Neosartorya pseudofischeri (strain number 4) and 1 Eurotium repens (strain number 17).
Touchdown PCR
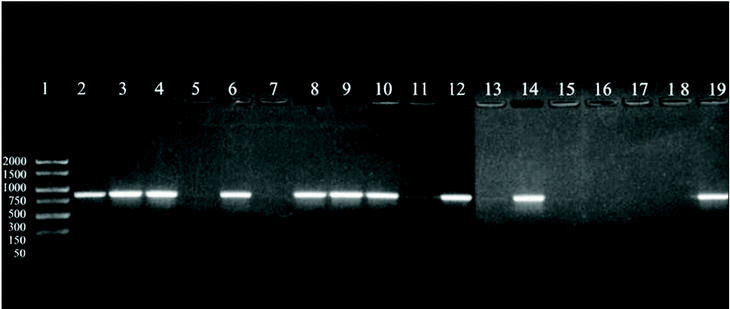
A 864 bp amplification product was obtained with the touchdown PCR protocol using Z19-276/Z19-660 primer pair from fungal strains number 1, 2, 3, 5, 7, 8, 9, 11, 13, and A. fumigatus from ATCC according, exactly, to the strains identified as A. fumigatus by phenotypical methods. No amplification band was obtained for the other strains which were morphologically classified as other different species than A. fumigatus (figure 1).
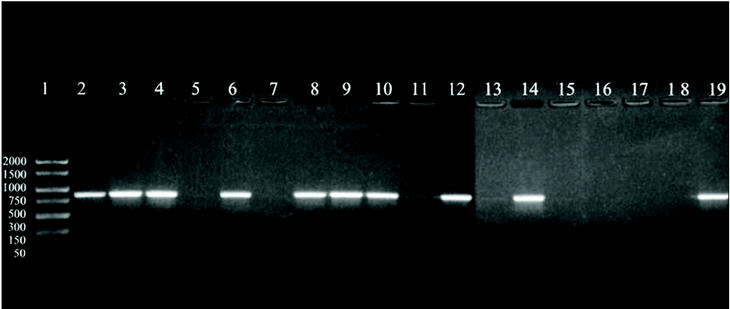
Figure 1. DNA from clinical and environmental fungal isolates amplified with primers Z19-276 and Z19-660 by touchdown protocol. Lanes: 1, PCR marker (base pair); 2, 3, 4, 6:A. fumigatus from clinical samples (strains nº 1, 2, 3, 5); 8, 9, 10, 12, 14:A. fumigatus from environmental samples (strains nº 7, 8, 9, 11, 13); 11: E. quadrilineata (strain nº 10), 13: A. niger, (ATCC1004) (strain nº 12); 15, 17:A. niger de origen ambiental (cepas nº 14, 16); 16:E. nidulans (cepa nº 15); 18: E. repens (cepa nº 17); 19:A. fumigatus from ATCC.
RAPD analysis
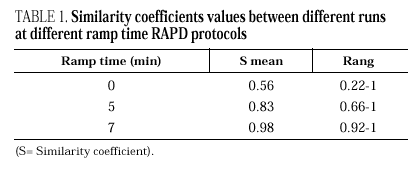
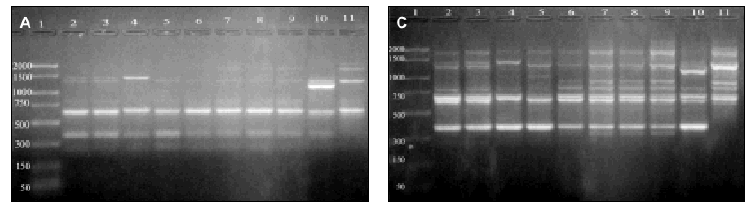
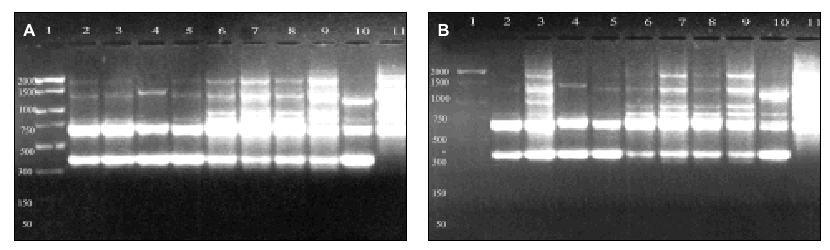
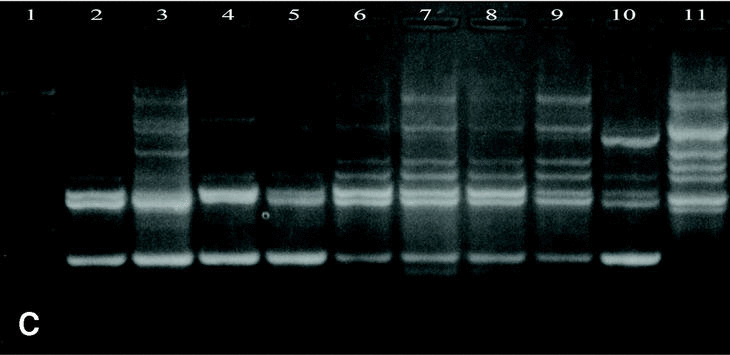
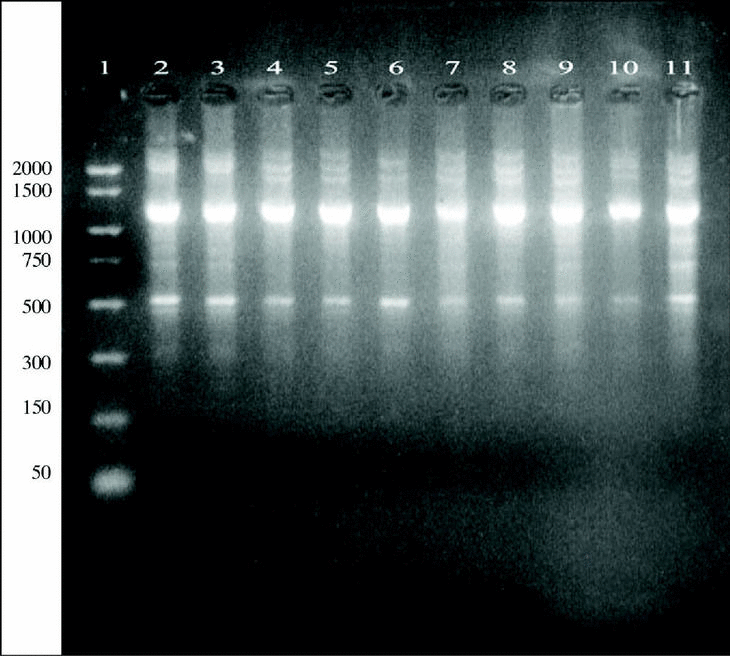
DNAs from a set of isolates comprising clinical and environmental A. fumigatus strains were screened with OPZ19 and R108 primers. The most discriminatory power was obtained for R108 primer which rendered six different types of patterns of bands (figure 2). The reproducibility and number of bands in RAPD analysis with R108 primer increased prolonging ramp times (table 1, figure 3). On the contrary, RAPD with OPZ19 primer had an acceptable reproducibility but very little resolution showing two different pattern of bands only, at any of the tested assay conditions (figure 4).
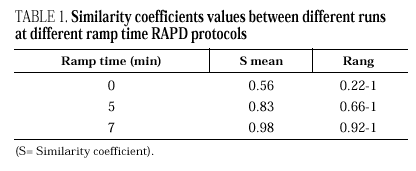
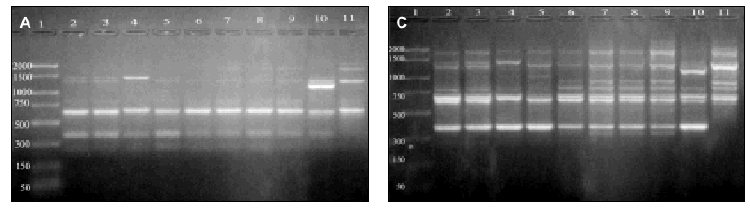
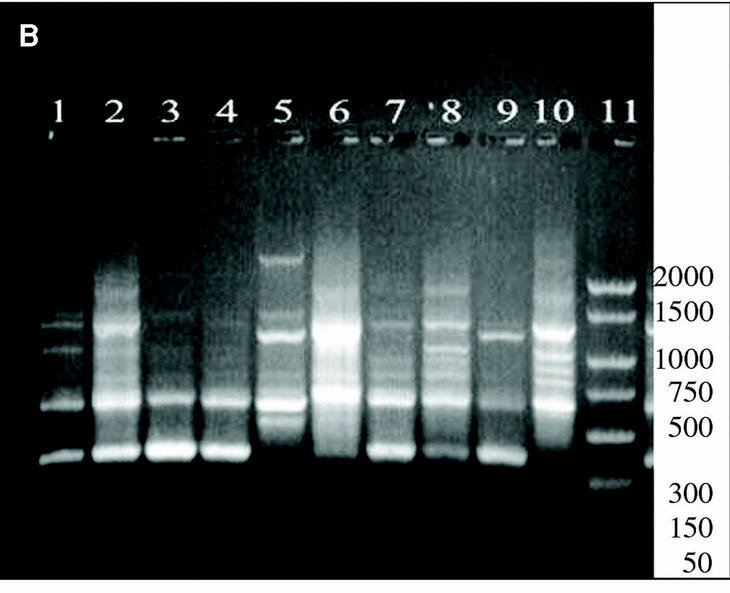
Figure 2. RAPD patterns produced with R108 primer. A: Ramp time = 0 minutes. Lanes: 1: PCR marker (base pair), lanes 2-10:A. fumigatus (strains nº 1, 2, 3, 5, 7, 8, 9, 11, 13), 11:A. fumigatus from ATCC. B: Ramp time = 5 minutes. Lanes: 1-9: A. fumigatus (strain nº 1, 2, 3, 5, 7, 8, 9, 11, 13); 10:A. fumigatus from ATCC; 11: Size marker (bp). C: Ramp time = 7 minutes. Lanes distribution as in figure 2 A.
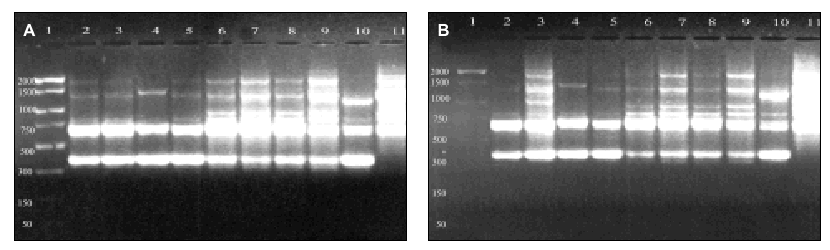
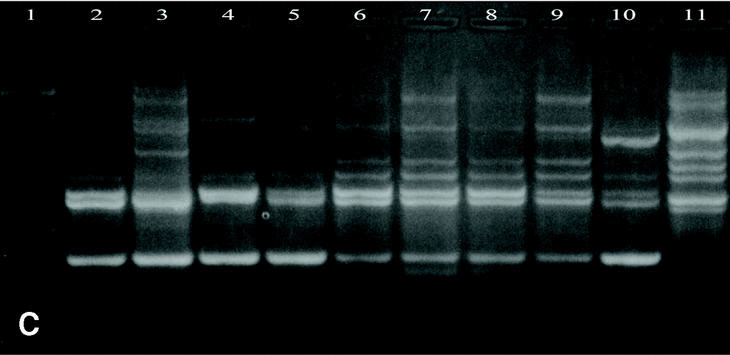
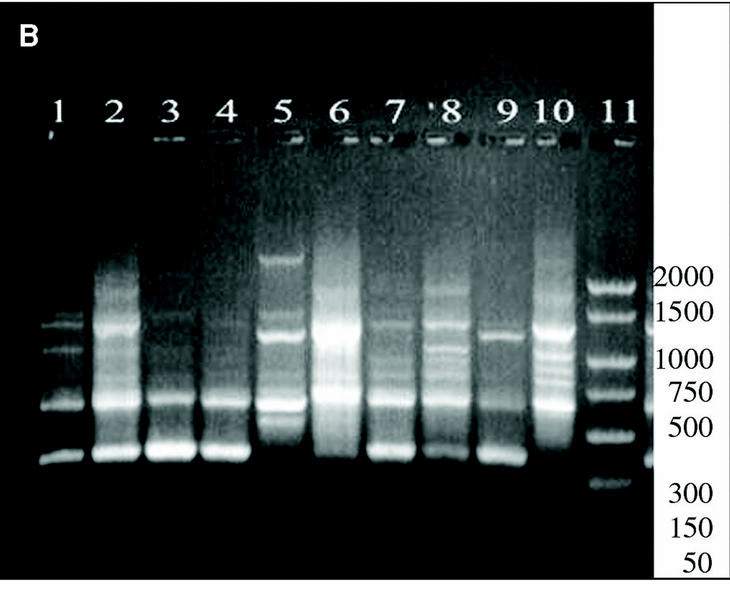
Figure 3. Patterns of bands obtained in different runs with different DNA extractions. A and B: Two different runs using the first DNA extraction. Figures C and D: Two different runs using the second DNA extraction. Lanes: 1: Size marker; 2-10:A. fumigatus (strain nº 1, 2, 3, 5, 7, 8, 9, 11, 13), 11:A. fumigatus from ATCC.
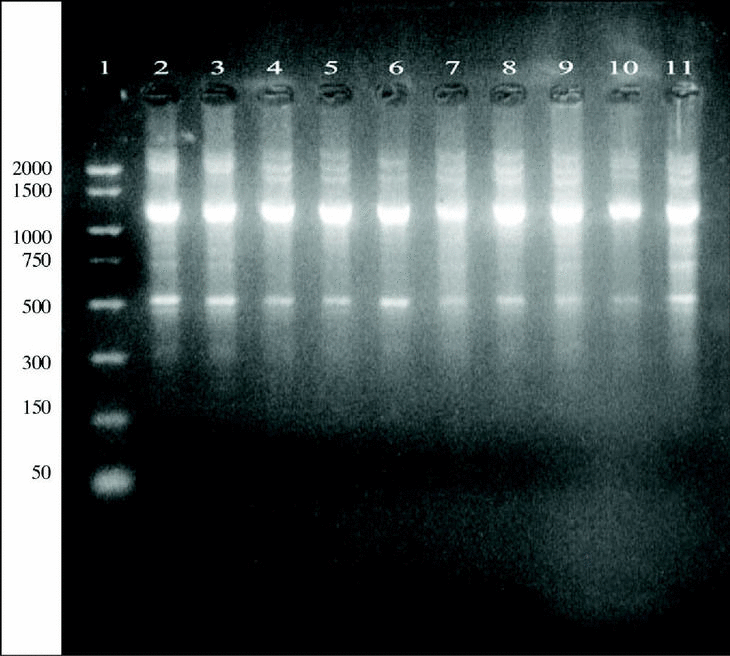
Figure 4. DNA from A. fumigatus strains amplified by RAPD-PCR using the OPZ-19 primer. Lanes: 1: PCR marker (bp); 2-10:A. fumigatus (strains number 1, 2, 3, 5, 7, 8, 9, 11, 13); 11: A. fumigatus from ATCC.
Discussion
By using touchdown annealing conditions, the Z19-276/660 primer-probe combination successfully amplified and detected DNAs from all the strains that were morphologically identified as A. fumigatus. So, this result confirms the great utility for accurate and timely identification of A. fumigatus from clinical and environmental samples. This detection is extremily important for the diagnosis and management of the diseases as well as for surveillance and epidemiologic studies. However, some uncommon varieties as A. fumigatus var. sclerotium and non pigmented strains may show faint or no reactivity under these conditions7.
Among several techniques that have been applied successfully to study fungal epidemiology, RAPD analysis is the most technically simple and often detects variation between isolates that are invariant with RFLP analysis10-12. However, its poor reproducibility has been considered to be a serious disadvantage. R108, a primer used successfully in RAPD analysis of A. fumigatus by other authors4,13, demonstrated a high discriminatory power, which is consistent with the results obtained by other authors4,14. Only two clinical strains were not distinguished by R108 protocol. These ones were from patients who stayed at the same operating theater and postsurgery care floor, at the same time. Therefore, they were, probably, infected by the same strain.
Although it is advisable that RAPD analysis be made with several primers, the use, only, of one of them with high discriminatory power, may be enough to manage the outbreak, avoiding more assays money and time-consuming15. In our study, prolonged ramp times between the annealing and the extension temperatures lead to an important increase in reproducibility of the patterns of bands obtained with R108 primer, according to the results obtained by P. Ellinghaus et al. with Candida sp. genome6. Also, the number, yield and reproducibility of the DNA fingerprint obtained was improved, significantly. A potential mechanism might be that the lower heating rates stabilize the primer/template complexes by avoiding premature detachment of the primer from the template. Otherwise, RAPD with OPZ19, a primer reactive with all strains of A. fumigatus, except non pigmented variants, had acceptable repetitivity with all the ramp times assayed, perhaps related with their own sequence because it is known that reproducibility and intensity of the bands in a fingerprint should be a function of several parameters including primer length and primer sequence16. However, prolonged ramp times reported an increase in number and yield of the DNA bands obtained.
In conclusion, RAPD analysis is a truly rapid and reliable tool in DNA fingerprinting. Patterns may be easier to repeat and interpret when drastically prolonged ramp times between annealing and extension are used. Combining touchdown PCR and RAPD analysis is a sensible and accurate method for epidemiologic studies of clinical outbreaks of A. fumigatus making use of the habitual techniques available in a current clinical microbiology laboratory.
Acknowledgments
We thank A.M. Commons for translation review of the manuscript.

