






Six out of ten completely sequenced bacterial genomes are pathogenic or opportunistic bacteria. The genome sequence of at least one strain of all the principal pathogenic bacteria will soon be available. This information should enable us to identify genes that encode virulence factors. As these genes are potential targets for drugs and vaccines, their identification should have considerable repercussions on prevention, diagnosis, and treatment of the main bacterial infectious diseases. Comparison of genome sequences of several strains of the same species should allow identification of the genetic clues responsible for the differing behavior of related bacterial pathogens. This article reviews the genomes from pathogenic bacteria that have been or are currently being sequenced, describes the main tasks to be accomplished after a genome sequence becomes available, and discusses the benefits of having the genome sequence of bacterial pathogens.
Seis de cada 10 genomas bacterianos cuya secuenciación se ha completado son de bacterias patógenas o que causan infecciones oportunistas. Muy pronto estarán disponibles las secuencias de los genomas de al menos una cepa de cada una de las principales bacterias patógenas. Esta información tendría que permitirnos identificar los genes que codifican factores de virulencia. Al ser dichos genes dianas potenciales para desarrollar fármacos y vacunas, su identificación debería tener considerables repercusiones en el diagnóstico, prevención y tratamiento de las principales infecciones bacterianas conocidas. La comparación de secuencias genómicas de las diversas cepas de una misma especie, tendría queposibilitar la identificación de las claves genéticas responsables de la diferente condcta de aquellos microorganismos patógenos bacterianos relacionados entre sí. Este artículo revisa cuáles son los genomas bacterianos que han sido ya secuenciados o lo están siendo en el momento actual. El artículo también describe qué tareas han de llevarse a cabo cuando se ha obtenido la secuencia completa de un genoma y analiza los beneficios de disponer de la secuencia genómica de bacterias patógenas
Most of our knowledge on the organization and dynamics of bacterial genomes has been obtained after years of sequencing individual genes or genome fragments. We have now entered the "genomic era"1, in which the task of sequencing a complete bacterial genome is becoming easier. A useful image to illustrate the impact of genomic research is to imagine a building firmly resting on Genome Projects, with three floors that represent the impact of genomics on biology, health, and society, respectively2. Seen in this way, genomics is a central discipline of biomedical research. The information extracted from genome projects will enable us to convert genome-based knowledge into health benefits and help to develop powerful new therapeutic and preventive approaches to infectious diseases2. This article reviews the genomes from pathogenic bacteria that have been or are currently being sequenced, briefly explains what "sequencing a genome" means, describes the main tasks to be accomplished after the genome sequence is obtained, and discusses the benefits of obtaining the complete sequence of bacterial pathogens.
Organization and dynamics of bacterial genomesBacterial genomes usually consist of a single circular chromosome between 0.5 and 10 megabases (Mb) in size that contains a unique origin and terminus of replication3. There are exceptions, however, such as linear chromosomes and bacteria that posses two or more chromosomes. Among pathogenic bacteria, Vibrio, Burkholderia, Leptospira and Brucella species are those with two or more chromosomes. Certain bacteria can also present one or more plasmids of varying length, which contain genes that may confer an advantage to the bacteria bearing them4. In some cases, the difference between a megaplasmid and a second chromosome may not be clear4.
The nucleotide composition of bacterial genomes varies greatly between species. Although the G + C (guanine-cytosine) content may vary locally within a genome, it is relatively uniform within a bacterial genus or species and ranges from around 25% in Mycoplasma to around 75% in some Micrococcus species. Although there is some heterogeneity in codon usage among genes in a genome (eg, depending on the expression level), Grantham et al5 proposed a genome hypothesis stating that genes in a given genome use the same coding strategy to choose among synonymous codons. That is, the G + C content and bias in codon usage is species-specific. Gene content and gene order are generally well preserved at close phylogenetic distances, but rapidly become less conserved among more distantly related organisms6. Prokaryotic genomes are not simply a random succession of genes, however, and there is selective pressure to maintain a certain genomic architecture7. Therefore, several elements, such as various genes, tRNAs and rRNAs, are usually found together or in a specific position7.
Bacterial genomes are dynamic. They are exposed to point mutations, duplications, inversions, transpositions, recombinations, insertions, and deletions, which can change them and influence their survival, lifestyle, and metabolic capabilities. Gene acquisition, also called horizontal gene transfer (HGT), may be the mechanism having the greatest impact on the organism's lifestyle, by conferring a novel metabolic capacity8,9. Although the fact that species are able to acquire DNA was discovered at the same time that DNA was identified as the genetic material10, HGT has been considered a rare event. However, when the first complete genome sequences became available, it was suggested that HGT might be more common than expected11,12. The length of bacterial genomes cannot increase indefinitely; hence, if genomes acquire genes, they must also lose them13.
Pathogenic species with a narrow range habitat, like the intracellular pathogen Mycoplasma and some other pathogenic bacteria, have fewer opportunities to acquire genes and show the lowest percentages of horizontally transferred genes11. Despite these low percentages, HGT has played a significant role in the evolution of pathogenic bacteria14, by the acquisition of antibiotic resistance genes15 and virulence factors16, for example. HGT, however, is not the only mechanism leading to increased pathogenicity. There are notable examples in which the increased pathogenicity of a strain results from modification of some genes or even from gene loss17. In other cases (eg, Pseudomonas aeruginosa lung infection), pathogenic bacteria can increase their mutation rate by error-prone repair of DNA mismatches in the process of adapting to new environments18. In some pathogenic bacteria (eg, Neisseria gonorrhoeae), programmed genomic changes involving site-specific recombination systems are induced, causing an antigenic phase variation in cell surface-expressed genes18.
The predominant evolutionary process in pathogenic bacteria is genome reduction17, resulting from a high rate of gene inactivation and gene loss, and a low rate of horizontal gene transfer. Thus, pathogenic and symbiotic bacteria have the shortest genomes, although some pathogens can retain long genomes. An extreme case of reductive evolution is illustrated by Mycobacterium leprae. Although the genome of M. leprae is 3.27 Mb in size (a large genome as compared to other pathogenic bacteria), it is under extensive gene inactivation since it contains 1,116 pseudogenes19. Pathogenic bacteria lose genes whose functions are no longer required in highly specialized niches. In comparison with non-pathogenic species, the genomes of pathogenic bacteria are more flexible7. Nonetheless, there are some differences between pathogens. Intracellular pathogens, such as Chlamydia, Spirochetes or Rickettsia species show low rearrangement rates and repetitive elements, whereas other pathogens have a high frequency of rearrangements and repeated, mobile elements, probably because of the need for fast adaptation and relaxed organizational constraints20.
The three phases of a genome project: sequencing, annotation and use of the dataSequencing the complete genome of bacteria is an interdisciplinary task that includes cloning the genome in fragments, obtaining its sequence, and annotating and analyzing it. These phases are explained below in more detail.
Phase 1: Genome sequencingSince the development of methods for DNA and protein sequencing in the late 1970s and early 1980s, the number of DNA and protein sequences in public databases has increased rapidly. These sequences include small complete genomes, such as those of bacteriophages, viruses, and certain organelles. The sequence of individual genes and small portions of genomes allowed a better understanding and characterization of microbial pathogens, and had considerable repercussions on biomedicine. Obtaining the complete sequence of a bacterium, however, was not feasible because of economic and technical limitations. This panorama changed when sequencing became cheaper, faster and more automatic.
The history of sequencing complete bacterial genomes is closely linked to the Human Genome Project (HGP). Since the beginnings of the HGP in 1990, one of the goals has been to obtain the complete genomic sequences of model laboratory organisms, such as the bacterium Escherichia coli. Complete sequencing of bacterial genomes in itself has substantial interest, however. The genome sequence provides the most fundamental information about an organism and yields clues to its evolution. The impulse the HGP gave to the field of genomics and the development of automated DNA sequencing machines made it possible to sequence the entire genome of an organism.
Although it was not considered a model organism, Haemophilus influenzae Rd was, in 1995, the first microbial genome reported in the literature21. It was sequenced by The Institute for Genomic Research (TIGR), in part as proof of a new whole-genome sequencing method called the shotgun method. Briefly, this technique consists of obtaining random fragments of the genome to be sequenced, cloning and sequencing these fragments, and computationally assembling the sequences obtained22. Because the assembly process is based on finding regions that overlap, more than 1 million bases must be sequenced in order to sequence a 1-Mb genome. The mean value of the number of times each base is sequenced in a genome project is called genome coverage and is usually between 6 and 822.
Although H. influenzae Rd was the first microbial genome to be sequenced, it was not the first microbial genome project initiated. In 1990 a project for sequencing the E. coli genome began. However, the initial strategy of this project was different from the shotgun approach. This strategy, called the clone contig approach, involves constructing a set of minimally overlapping clones whose relative position is known (some kind of map is therefore needed) and sequencing only the fragments that are known to overlap. The clone contig strategy was initially proposed for sequencing the human genome by the HGP and is the conventional method used to obtain the sequence of eukaryotic genomes. From 1992 to 1995, a total of 1.92 Mb of the E. coli genome was sequenced using this strategy. The success of the shotgun approach for sequencing the H. influenzae genome led to changes in the strategy for sequencing E. coli and the project was concluded using a shotgun strategy. The complete genome of E. coli K12 was finally published in 199723.
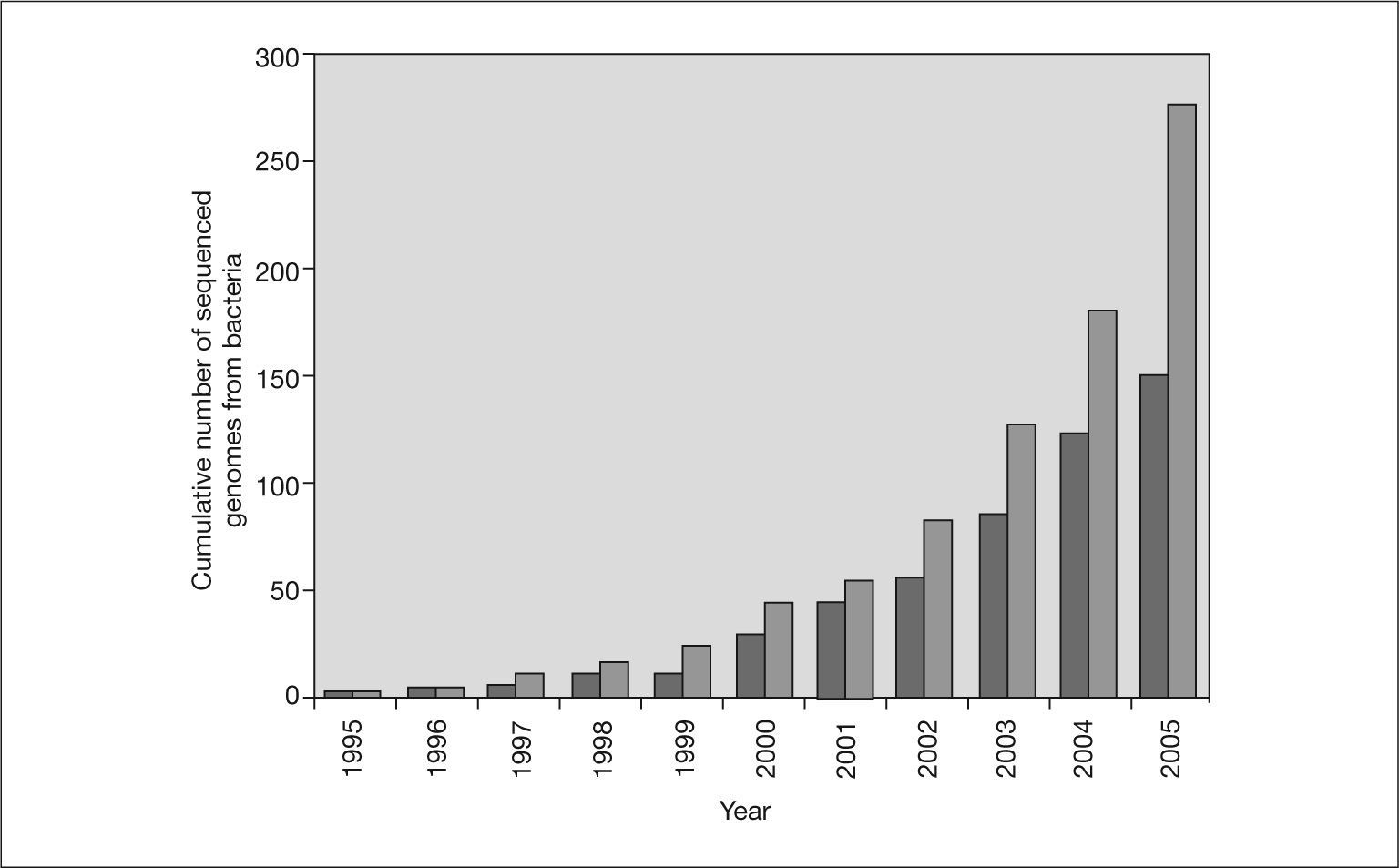
Since that time, with few exceptions, complete bacterial genomes have been sequenced using the shotgun strategy. The cumulative number of bacterial genomes reported since 1995 is shown in figure 1. The number of complete sequenced genomes has increased rapidly, in parallel to the growth of sequence databases, such as GenBank. This trend will continue in the next years since there are more than 500 bacterial genome projects in progress. The next step is to have the complete sequence of 1000 genomes. This will be possible in the near future thanks to advances in DNA sequencers and the development of new sequencing technologies. One of the next-generation DNA sequencers has abandoned traditional Sanger chemistry for an on-bead sequencing-by-synthesis approach to generate more than 25 million bases, at 99% or better accuracy, in one four-hour run24. This represents an approximately 100-fold increase in throughput over current Sanger sequencing technology and has allowed the complete genome of Mycoplasmagenitalium to be sequenced in only four hours24. Claire Fraser's team at the Institute for Genomic Research took three months to work out the sequence of this microorganism in 199525.
. Light gray bars indicate the total number of bacterial genomes and dark gray bars indicate only genomes from human or animal pathogens.")
Cumulative number of published bacterial genomes. The data are from the Genome Project Database at NCBI (http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db = genomeprj). Light gray bars indicate the total number of bacterial genomes and dark gray bars indicate only genomes from human or animal pathogens.
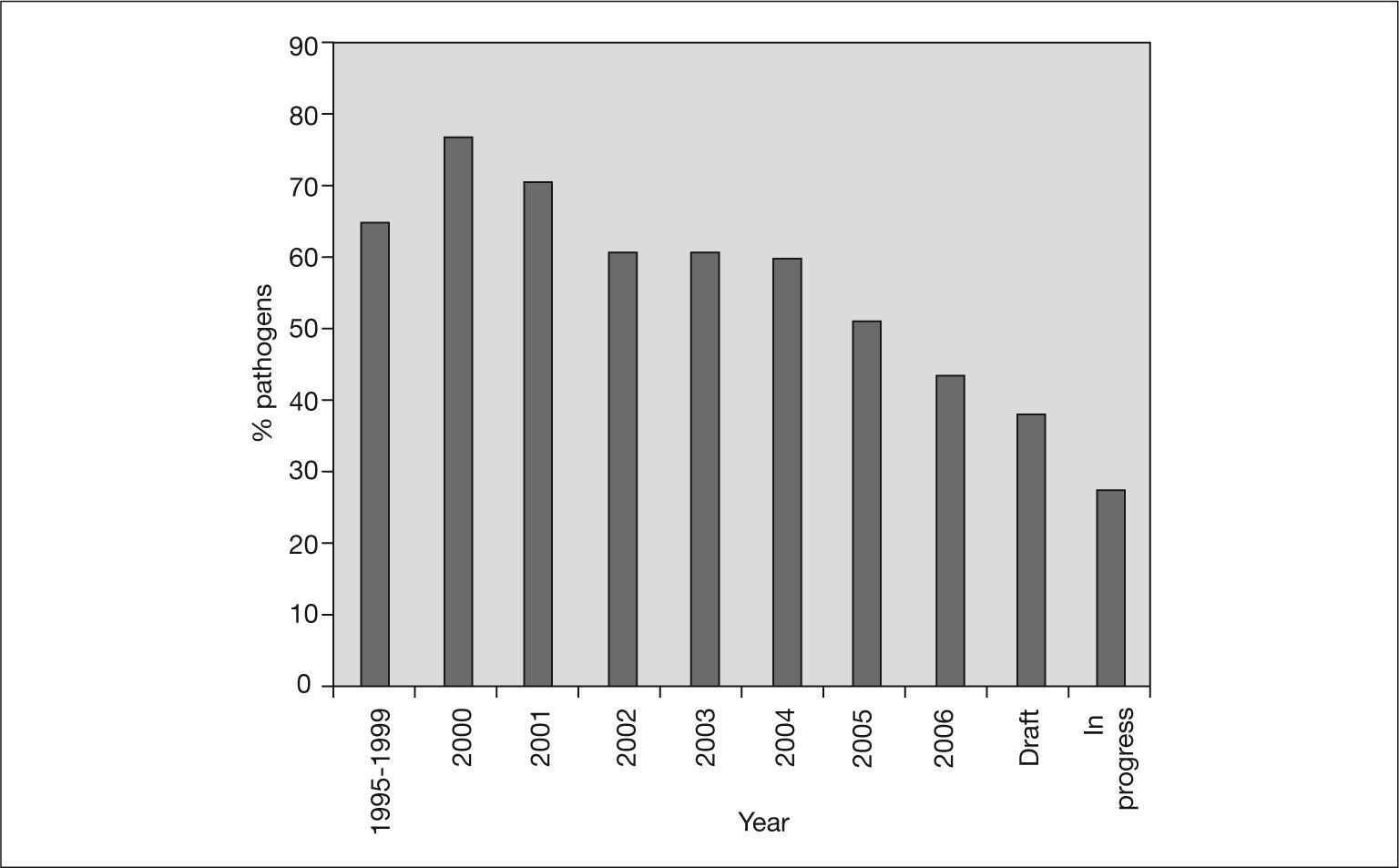
Infectious diseases are the leading cause of mortality in the population under 40 years. It is not surprising, therefore, that the genomes of an increasing number of bacterial pathogens have been sequenced. These sequences are an important tool for gaining knowledge of the genetic makeup of these pathogens and insight into how they may be controlled26. Figure 1 also shows the cumulative number of published genomes from human pathogenic bacteria. Nearly 60% of all the bacterial genomes sequenced are from pathogenic bacteria. However, this percentage has not always been the same (fig. 2). In the first years of genome sequencing, more than 70% of complete bacterial genomes were from pathogens. However, several institutions soon focused on nonpathogenic bacteria. With funds from the US Department of Energy (DOE) and other institutions, the genomes of the cyanobacterium Synechocystis sp. and the archaea Methanococcus jannaschii, Methanothermobacter thermautotrophicus and Archaeoglobus fulgidus were sequenced in 1996 and 1997. Interest in sequencing the genomes of nonpathogenic bacteria has increased, and the percentage of pathogen genomes now being sequenced is only 36% for drafted genome projects (those in which the sequencing task has concluded and sequences are being assembled and annotated) and 28% for projects in progress (those still in the sequencing phase) (fig. 2). Apart from biomedical applications, there are many other reasons for sequencing bacterial genomes. For example, biotechnological purposes (eg, sequencing bacteria used in bioremediation or wastewater treatment, bacteria of interest for agriculture or energy production, and bacteria to produce chemical compounds, such as amino acids or vitamin C), environmental efforts (eg, sequencing photosynthetic or extremophilic bacteria), and evolutionary reasons27.

Yearly percentages of bacterial genomes that have been completely sequenced and correspond to a pathogenic or opportunistic organism. Interest in sequencing the genome of non-pathogenic bacteria is increasing. Draft refers to genomes whose sequencing task is finished and sequences are being assembled and annotated. In progress refers to genomes that are still at the sequencing phase.
A comprehensive list of completed and ongoing pathogenic bacterial genome projects is presented in table 1. The species in the table are classified according to the Gram staining method and the standards of a medical microbiology textbook. The table shows that the most important human bacterial pathogens have been or are now being sequenced. Thus, the genome sequences of pathogenic enterobacteria, intracellular pathogens, low G + C gram-positive organisms, actinobacteria and other high G + C gram-positive bacteria are available. Focusing on the diseases these pathogens cause, we see that the most important infectious diseases, such as tuberculosis, leprosy, meningitis, cholera, staphylococcal and streptococcal infections, trachoma, and syphilis are represented. In addition, many pathogens have been sequenced more than once, with sequencing of several strains. Helicobacter pylori was the first case in which the genome sequences of two different strains of the same bacteria were obtained28. The main reason why this occurred was a genomic race between a non-profit organization and a pharmaceutical company to be the first to sequence this genome, although the pathogenicity of the two strains is significantly different (the 26695 strain was originally isolated from a patient with gastritis and the J99 strain from a patient with duodenal ulcer).
Pathogenic bacterial genome sequencing projects completed or in progress
| Family Genus, species | Disease | Complete strains | Draft/In progress strains* | Genome length (Mb) | G + C content |
| Gram-positive cocci | |||||
| Staphylococcaceae | |||||
| Staphylococcus aureus | Pyogenic infections, toxicosis | 9 | 2/0 | 2.7-2.8 | 32 |
| S. epidermidis | Opportunist infections | 2 | 0/0 | 2.5-2.6 | 32 |
| S. saprophyticus | Acute urinary tract infections | 1 | 0/0 | 2.5 | 33 |
| S. haemolyticus | Opportunistic pathogen of immunocompromised patients | 1 | 0/0 | 2.7 | 32 |
| Streptococcaceae | |||||
| Streptococcus pyogenes | Tonsillitis, scarlet fever, skin infections | 11 | 1/1 | 1.9 | 38 |
| S. pneumoniae | Pneumonia, otitis media, sinusitis | 2 | 1/2 | 1.8-2.2 | 39 |
| S. agalactiae | Neonatal sepsis and meningitis | 3 | 5/0 | 2.1-2.2 | 35-36 |
| S. mutans | Caries | 1 | 0/0 | 2 | 36 |
| S. gordonii | Caries, periodontal disease and endocarditis | 0 | 0/1 | 2 | na |
| S. mitis | Endocarditis | 0 | 0/1 | na | na |
| S. salivarius | Endocarditis, blood infection, and peritonitis | 0 | 0/2 | na | na |
| S. sanguinis | Caries, periodontal disease and endocarditis | 0 | 0/1 | na | na |
| S. sobrinus | Caries and endocarditis | 0 | 0/1 | na | na |
| Enterocococcaceae | |||||
| Enterococcus faecalis | Opportunistic infections | 1 | 0/0 | 3.2 | 37 |
| E. faecium | Opportunistic infections | 0 | 1/0 | 2.8 | 37 |
| Endospore-forming Gram-positive rods | |||||
| Bacillaceae | |||||
| Bacillus anthracis | Anthrax | 3 | 7/0 | 5-5.4 | 35 |
| B. cereus | Food poisoning | 3 | 2/2 | 5.2-5.3 | 35 |
| B. licheniformis | Food poisoning | 2 | 0/0 | 4.3 | 46 |
| Clostridiaceae | |||||
| Clostridium tetani | Tetanus | 1 | 0/0 | 2.8 | 28 |
| C. perfringens | Gas gangrene | 1 | 0/2 | 3 | 28 |
| C. difficile | Pseudomembranose colitis | 0 | 1/1 | 3.9 | 28 |
| C. botulinum | Botulism | 0 | 0/1 | na | na |
| Regular non-sporing Gram-positive rods | |||||
| Listeria monocytogenes | Opportunistic food-born diseases | 2 | 2/0 | 2.9 | 37 |
| Irregular non-sporing Gram-positive rods | |||||
| Corynebacteriaceae | |||||
| Corynebacterium diphtheriae | Diphtheria | 1 | 0/0 | 2.5 | 53 |
| C. jeikeium | Endocarditis and nosocomial infections | 1 | 0/0 | 2.5 | 61 |
| Actinomycetaceae | |||||
| Actinomyces naeslundii | Actinomycosis and gingivitis | 0 | 0/1 | na | na |
| Nocardiaceae | |||||
| Nocardia farcinica | Nocardiosis | 1 | 0/0 | 6 | 70 |
| Propionibacteriaceae | |||||
| Propionibacterium acnes | Acne | 1 | 0/0 | 2.6 | 60 |
| Cellulomonadaceae | |||||
| Tropheryma whipplei | Whipple's disease | 2 | 0/0 | 0.93 | 46 |
| Mycobacteria | |||||
| Mycobacteriaceae | |||||
| Mycobacterium tuberculosis | Tuberculosis | 2 | 2/1 | 4.4 | 65 |
| M. leprae | Leprosy | 1 | 0/0 | 3.2 | 57 |
| M. abscessus | Lung, skin, and wound infections | 0 | 0/1 | na | na |
| M. bovis | Peritoneal tuberculosis | 1 | 0/1 | 4.3 | 65 |
| M. ulcerans | Buruli ulcer | 0 | 0/1 | na | na |
| M. avium | Disseminated infections in immunocompromized humans | 1 | 0/1 | 4.8 | 69 |
| M. chelonae | Wound, cornea, and skin infections | 0 | 0/1 | na | na |
| M. smegmatis | Soft tissue lesions and bacteremia | 0 | 0/1 | na | na |
| Gram-negative aerobic cocci and coccobacilli | |||||
| Neisseriaceae | |||||
| Neisseria gonorrheae | Gonorrhea | 1 | 0/0 | 2.1 | 52 |
| N. meningitidis | Meningitis/sepsis | 2 | 0/1 | 2.2-2.3 | 51 |
| Moraxellaceae | |||||
| Moraxella catarrhalis | Respiratory infections | 0 | 0/1 | na | na |
| Acinetobacter calcoaceticus | Nosocomial pathogen | 1 | 0/0 | 3.6 | 40 |
| A. baumannii | Nosocomial pathogen | 0 | 0/2 | na | na |
| Gram-negative facultative anaerobic rods | |||||
| Enterobacteriaceae | |||||
| Escherichia coli | Gut diseases, nosocomial infections | 6 | 9/2 | 4.6-5.5 | 50 |
| Salmonella typhi, S. paratyphi | Typhoid/paratyphoid fever | 4 | 0/2 | 4.6-4.8 | 52 |
| Salmonella typhimurium | Gastroenteritis | 1 | 0/3 | 4.9 | 52 |
| Shigella dysenteriae | Bacterial dysentery | 1 | 1/1 | 4.3 | 50 |
| Shigella flexneri, S. boydii, S. sonnei | Bacterial dysentery | 4 | 1/3 | 4.5-4.6 | 51 |
| Klebsiella, Enterobacter, Citrobacter, Proteus, Serratia, Morganella, Providencia | Opportunistic pathogens | 0 | 0/6 | na | na |
| Yersinia pestis | Bubonic plague, pulmonary plague | 3 | 1/2 | 4.5-4.6 | 47 |
| Y. enterocolitica | Enteritis, lymphadenitis | na | 0/1 | na | na |
| Y. pseudotuberculosis | Tuberculosis-like disease | 1 | 1/0 | 4.7 | 47 |
| Vibrionaceae | |||||
| Vibrio cholerae | Cholera | 1 | 5/0 | 4** | 47 |
| V. parahaemolyticus | Seafood-associated food poisoning | 1 | 0/0 | 5.1** | 45 |
| V. vulnificus | Wound infections, gastroenteritis, primary septicemia | 2 | 0/0 | 5** | 46 |
| Pasteurellaceae | |||||
| Pasteurella multocida | Opportunistic pathogen | 1 | 0/0 | 2.2 | 40 |
| Haemophilus influenzae | Meningitis, respiratory tract infections | 2 | 2/11 | 1.8-1.9 | 38 |
| H. ducrey | Sexually-transmitted chancroid | 1 | 0/0 | 1.7 | 38 |
| H. somnus | Pneumonia, arthritis, myocarditis, reproductive problems | 0 | 2/0 | 2.2 | 37 |
| Actinobacillus actinomycetemcomitans | Periodontal infections | 0 | 1/0 | na | na |
| Shewanellaceae | |||||
| Shewanella putrefaciens | Ears and soft infections, bacteremia, meningitis | 0 | 1/2 | na | na |
| Gram-negative aerobic rods | |||||
| Pseudomonadaceae | |||||
| Pseudomonas aeruginosa | Opportunistic infections | 1 | 4/0 | 6.2 | 66 |
| Burkholderiaceae | |||||
| Burkholderia mallei | Skin abscesses | 1 | 8/0 | 5.3-6** | 68 |
| B. pseudomallei | Melioidosis | 2 | 8/0 | 7.1-7.4** | 67 |
| Burkholderia sp. | Necrotizing pneumonia, chronic infections | 1 | 0/0 | 8.7** | 66 |
| B. cenocepacia | Opportunistic infection of cystic fibrosis patients | na | 3/1 | 7-8** | 66 |
| B. dolosa | Necrotizing pneumonia, chronic infections | na | 1/0 | 6.2** | 66 |
| B. vietnamiensis | Necrotizing pneumonia, chronic infections | na | 1/0 | 8.4** | 65 |
| B. xenovorans | Opportunistic infection of cystic fibrosis patients | 1 | 0/1 | 9.8** | 62 |
| Legionellaceae | |||||
| Legionella pneumophila | Legionellosis | 3 | na | 3.3-3.5 | |
| L. longbeachae | Legionellosis in Australia | na | 0/1 | na | na |
| L. hackeliae | Pneumonia, respiratory infections | 0 | 0/1 | na | na |
| Brucellaceae | |||||
| Brucella melitensis | Brucellosis | 2 | 0/1 | 3.3** | 57 |
| B. abortus | Brucellosis | 1 | 0/0 | 3.3** | 57 |
| B. suis | Brucellosis | 1 | 0/0 | 3.3** | 57 |
| Ochrobactrum anthropi | Opportunistic infections | 0 | 0/1 | na | na |
| Alcaligenaceae | |||||
| Bordetella pertussis, B. parapertusis, B. bronchiseptica | Bronchitis and other respiratory diseases | 3 | 0/0 | 4.1 | 67 |
| Francisellaceae | |||||
| Francisella tularensis | Tularemia | 2 | 0/2 | 1.8 | 32 |
| F. philomiragia | Pneumonia and septicemia | 0 | 0/1 | na | na |
| Gram-negative rods, straight, curved, and helical, strictly anaerobic | |||||
| Bacteroidaceae | |||||
| Bacteroides fragilis | Opportunistic pathogen of the intestinal tract | 2 | 0/0 | 5.2 | 43 |
| Tannerella forsythensis | Progression of periodontal disease | 0 | 0/2 | na | na |
| Porphyromonadaceae | |||||
| Porphyromonas gingivalis | Periodontal disease | 1 | 0/0 | 2.3 | 48 |
| Fusobacteriaceae | |||||
| Fusobacterium nucleatum | Tooth decay | 1 | 1/0 | 2.1 | 27 |
| Prevotellaceae | |||||
| Prevotella intermedia | Necrotizing periodontal disease | 0 | 0/1 | na | na |
| P. ruminicola | Necrotizing periodontal disease | 0 | 0/1 | na | na |
| Aerobic/microaerophilic, motile, helical/vibrioid Gram-negative rod bacteria | |||||
| Campylobacteriaceae | |||||
| Campylobacter jejuni | Food poisoning | 2 | 5/1 | 1.7 | 30 |
| C. fetus | Opportunistic infections: sepsis, endocarditis | 0 | 1/1 | 1.8 | 33 |
| C. lari | Gastroenteritis and bacteremia | 0 | 1/0 | na | na |
| C. upsaliensis | Bacteremia and septicemia in immunocompromised individuals | 0 | 1/0 | na | na |
| Helicobacteriaceae | |||||
| Helicobacter pylori | Gastric inflammation and peptic ulcer | 2 | 0/0 | 1.7 | 38-39 |
| The Spirochetes. Gram-negative, helical bacteria | |||||
| Spirochaetaceae | |||||
| Treponema pallidum | Syphilis | 1 | 0/0 | 1.3 | 52 |
| T. denticola | Periodontal disease, gum inflammation | 1 | 0/0 | 2.8 | 37 |
| Borrelia burgdorferi | Lyme disease | 1 | 0/0 | 0.9 | 28 |
| B. garinii | Tick-borne borreliosis in Europe | 1 | 0/0 | 0.9 | 28 |
| Leptospiraceae | |||||
| Leptospira interrogans | Leptospirosis | 2 | 0/0 | 4.7 | 35 |
| Rickettsiae, Coxiellae, Ehrlichiae, Bartonellae, and Chlamydiae | |||||
| Rickettsiaceae | |||||
| Rickettsia prowazekii | Louse-borne typhus and Mediterranean spotted fever | 1 | 0/0 | 1.1 | 29 |
| R. rickettsii | Rocky Mountain spotted fever | 0 | 1/0 | 1.2 | 32 |
| R. conorii | Rocky Mountain spotted fever | 1 | 0/0 | 1.2 | 32 |
| R. africae | African tick-bite fever | 0 | 0/1 | na | na |
| R. akari | Rickettsialpox disease | 0 | 1/0 | 1.2 | 32 |
| R. felis | Fleaborne spotted fever | 1 | 0/0 | 1.4 | 32 |
| R. massiliae | Spotted fever | 0 | 0/1 | na | na |
| R. sibirica | North Asian tick typhus | 0 | 1/0 | 1.2 | 32 |
| R. slovaca | Tickborne lymphadenopathy | 0 | 0/1 | na | na |
| R. typhi | Endemic typhus | 1 | 0/0 | 1.1 | 28 |
| Anaplasmataceae | |||||
| Neorickettsia sennetsu | Sennetsu fever | 1 | 0/0 | 0.9 | 41 |
| Coxelliaceae | |||||
| Coxiella burnetii | Q fever | 1 | 2/0 | 1.9 | 42 |
| Ehrlichiaceae | |||||
| Ehrlichia chaffeensis | Monocytic ehrlichiosis | 1 | 1/0 | 1-1.2 | 30 |
| Anaplasma phagocytophilum | Granulocytic anaplasmosis | 1 | 0/0 | 1.4 | 41 |
| Bartonellaceae | |||||
| Bartonella bacilliformis | Carrion's disease | 0 | 1/1 | 1.4 | 38 |
| B. henselae | Bacillary angiomatosis | 1 | 0/0 | 1.9 | 38 |
| B. quintana | 1 | 0/0 | 1.6 | 38 | |
| Chlamydiaceae | |||||
| Chlamydia trachomatis | Trachoma | 2 | 0/0 | 1.0 | 41 |
| Chlamydophila pneumoniae | Pharyngitis, bronchitis and pneumonitis | 4 | na | 1.2 | 40 |
| Chlamydophila psittaci | Psitacosis | 0 | 0/1 | na | na |
| Chlamydia felis | Pharyngitis, bronchitis and pneumonitis | 1 | 0/0 | 1.2 | 39 |
| Simkaniaceae | |||||
| Simkania negevensis | Pneumonia, bronchiolitis and chronic obstructive pulmonary disease | 0 | 0/1 | na | na |
| Mycoplasmas | |||||
| Mycoplasmataceae | |||||
| Mycoplasma pneumoniae | Tracheobronchitis and atypical pneumonia | 1 | na | 0.8 | 40 |
| M. genitalium | Urogenital and respiratory tract infections | 1 | 1/1 | 0.6 | 31 |
| M. fermentans | Respiratory illness and arthritis | 0 | 0/1 | na | na |
| M. penetrans | Urogenital or respiratory tract infections | 1 | 0/0 | 1.4 | 25 |
| Ureaplasma urealyticum | Urogenital or respiratory tracts infections | 1 | 0/0 | 0.8 | 25 |
The data are from the Genome Project Database at NCBI as of 22nd May 2006. Bacterial classification is taken from the Kayser et al53 Medical Microbiology textbook.
Nowadays, however, there are several projects for sequencing the genomes of different strains, mainly because of important differences between them (eg, in pathogenicity or antibiotic resistance). Comparison of the genome sequence of different strains for example, non-pathogenic and pathogenic strains will provide important clues as to which of the main genetic elements are responsible for infectious diseases and what evolutionary forces generate the differences between strains29.
In some cases, genomic variation between strains of the same species is minimal and subtle. For example, Bacillus anthracis strains vary by only a few single nucleotide polymorphisms (SNPs). In other cases, however, surprisingly high levels of diversity are evident. E. coli strains vary by as much as 25% of their genome30. Differences between genomes usually occur in the form of large genome islands that contribute to the acquisition of virulence factors or antibiotic resistance30. The high degree of intrastrain diversity sometimes observed suggests that a single genome is not representative of a species30. Therefore, an appropriate genomic view of bacterial populations suggests that, in addition to a ‘core’ set of genes found in all strains of a given species, strain-specific genes are also present, forming the so-called ‘auxiliary’ set. Both the core and auxiliary sets form the ‘species genome’, which comprises all the genes present in all strains of a given species31. Although nearly all the genomes of the main pathogenic bacteria have been sequenced or are in progress, some have not yet been included in any genome project. Pathogenic bacteria whose genomes have not yet been sequenced include several Nocardia species (for example, N. asteroids and N. brasiliensis) that cause nocardiosis, several Borrelia species that produce epidemic relapsing fever, Actinomyces israelii, an important bacterium responsible for actinomycosis, Gardnerella vaginalis, which produces vaginosis, Calymmatobacterium granulomatis, the pathogenic agent responsible for granuloma inguinale, Erysipelothrix rhusiopathiae, the etiologic agent of erysipeloid, and some species of the Neisseriaceae family that cause nosocomial infections.
Phase 2: Genome annotationKnowledge of a species' genome sequence does not directly tell us how this genetic information leads to the observable traits and behavior (phenotype) we wish to understand32. After a bacterial genome has been sequenced, the next phase is to annotate it. Annotation can be defined as a process by which structural and functional information is inferred for genes or proteins, usually on the basis of similarity to previously characterized sequences in public databases33. This task requires the use of several computational methods and has helped to develop the field of bioinformatics. Several bioinformatic tools must be executed to obtain the maximum information from a genome sequence. Without the development of bioinformatics, the complete genome sequence of an organism would be indecipherable.
The first task in analyzing a genome sequence is gene finding. In prokaryotic genomes this task is remarkably accurate, unlike in eukaryotic genomes. Gene finding in prokaryotic genomes is relatively simple because of the high gene density and absence of introns. Using modern bioinformatic tools, genes are annotated along with other structural parameters of the genome, such as the position of tRNAs and rRNAs and the origin and terminus of replication. The set of gene annotations obtained provides the basis for further analyses, such as prediction of the function of each gene. Gene function is generally predicted by database comparisons with similar genes of known function or similar genes with a predicted function. However, this procedure can introduce annotation errors that are very difficult to detect. Moreover, the function of a large percentage of genes (a third of all genes in some organisms) is unknown.
Some microbial genome projects are under constant revision33. For example, have a look at the GenoList genome browser at the Institute Pasteur (table 2). Several new annotation tools have also been developed34. However, some genome annotations have never been revisited. This is not a serious drawback if the final quality of the sequence is high, but several cases of erroneous annotation35,36, and perhaps a case of erroneous assembly of the sequenced fragments37, suggest that mistakes in genomic data may be more frequent than expected38,39. These aspects should be taken into consideration when a non-specialist in genomics uses publicly available genomic data.
Databases and tools related with bacterial genomic data
| NCBI Entrez Genome Project database: |
| http://www.ncbi.nlm.nih.gov/entrez/query.fcgi7db = genomeprj |
| A searchable collection of complete and incomplete (in-progress) large-scale sequencing, assembly, annotation, and mapping projects for cellular organisms |
| NCBI, Bacteria Genome Database: |
| http://www.ncbi.nlm.nih.gov/genomes/static/eub.html |
| The Genome database provides views for a variety of genomes, complete chromosomes, sequence maps with contigs, and integrated genetic and physical maps |
| Bacterial Genomes at The Sanger Institute: |
| http://www.sanger.ac.uk/Projects/Microbes/ |
| This web contains a list of funded, on-going, or completed projects of pathogens sequenced at this institute |
| TIGR Comprehensive Microbial Resource (CMR): |
| http://cmr.tigr.org/tigr-scripts/CMR/CmrHomePage.cgi |
| A free website displaying information on all the publicly available, complete prokaryotic genomes |
| GOLD: Genomes OnLine Database: |
| http://www.genomesonline.org/ |
| A genome database containing information about which genomes have been sequenced or are in progress |
| GenoList genome browser at Institute Pasteur: |
| http://genolist.pasteur.fr/ |
| Contains access to diverse genome browsers of pathogenic bacteria |
| Genome Information Broker: |
| http://gib.genes.nig.ac.jp/ |
| A comprehensive data repository of complete microbial genomes in the public domain. Many microbial genomes can be explored graphically |
| Microbial Genome Database for Comparative Analysis (MBGD): |
| http://mbgd.genome.ad.jp/ |
| A database for comparative analysis of completely sequenced microbial genomes |
| Virulence Factors of Bacterial Pathogens (VFDB): |
| http://zdsys.chgb.org.cn/VFs/main.htm |
| VFDB is an integrated and comprehensive database of virulence factors for bacterial pathogens |
| Islander, a Database of Genomic Islands: |
| http://www.indiana.edu/∼islander |
| This database contains genomic islands discovered in completely sequenced bacterial genomes |
| IslandPath: |
| http://www.pathogenomics.sfu.ca/islandpath/update/IPindex.pl |
| An aid to the identification of genomic islands, including pathogenicity islands, of potentially horizontally transferred genes |
| HGT-DB: |
| http://www.tinet.org/∼debb/HGT/ |
| A database containing the prediction of horizontally transferred genes in several prokaryotic complete genomes |
| E. coli genome project: |
| http://www.genome.wisc.edu |
| A site devoted to the E. coli genome project with an updated annotation of the genome |
Genome projects generate a large amount of data. Since release 149 of GenBank in August 2005, the number of bases from whole genome shotgun sequencing projects now exceeds the number of bases in the traditional Gen-Bank divisions. Just as important as obtaining and annotating the genome sequence of bacterial pathogens, is how this information is made available. Genome sequences are stored in public databases accessible over the Internet. To facilitate their use, several databases and tools are also available, but it is usually difficult to access genomic information for non-specialized research. Table 2 shows several databases and tools related with bacterial genomic data tht can be used to access this kind of information.
The genome sequence of an organism also generates new questions and stimulates new in silico and in vitro experiments. Some of the computational methods used to analyze the genomes of bacterial pathogens may help to identify virulence factors and pathogenicity islands, predict surface-exposed and secreted proteins, analyze metabolic pathways, identify phase-variable genes and antigenic sequences, and characterize human polymorphisms associated with infectious disease.
Phase 3: From whole genome sequence to applicationsAll the effort and money invested in determining the complete sequence of a bacterial pathogen would be wasted if the information obtained were not used in the prevention, diagnosis, or treatment of bacterial infectious diseases. The genome sequence of bacterial pathogens provides basic information for understanding the biology of bacteria and the pathogenesis of the diseases they cause. This information should help to identify genes and pathways that have a role in health and disease. It should also help in the development of genome-based diagnostic methods for predicting an individual's susceptibility to a disease and early detection of illness, as well as methods that catalyze the conversion of genomic information into therapeutic advances2. It is beyond the scope of this review to describe all the possible applications of genomic knowledge derived from the genome sequence of pathogenic bacteria. Genome sequences must be viewed as additional tools in our battle against infectious agents, and the use of these tools depends on the training, ability, and imagination of researchers. Below we describe just a few potential uses of microbial genome sequences. See Brinkman and Fueyo26, Raskin et al30, Subramanian et al40, and Weinstock et al41 for more.
Metabolic reconstruction and prediction of highly expressed genesOnce the genome of a bacterium has been sequenced and annotated, reconstruction of the encoded metabolic pathways is possible. Several databases contain the metabolic information derived from complete genomes42,43. As well as determining all the metabolic pathways present in a bacterium, it is also interesting to identify which genes and pathways are the most highly expressed. Estimation of the overall gene expression pattern of a pathogen is useful for determining the basic metabolic pathways most extensively used by the species and identifying which genes are involved in virulence and survival in host cells44. Highly expressed genes can be predicted experimentally or computationally, based on the finding that codon bias (the bias for using a particular set of synonymous codons rather than using synonymous codons at random) tends to be much stronger in highly expressed genes than in genes expressed at lower levels.
Whole genome DNA-microarraysIf we know the complete genome of a pathogenic bacterium, we can design whole-genome DNA microarrays. Briefly, microarrays consist of a series of nucleic acid targets immobilized on a solid substrate. Hybridization of fluorescently labeled probes to these targets enables analysis of the relative concentrations of mRNA or DNA in a sample45. Microarrays can be used in different ways to resolve different questions, usually with consequential improvements in the diagnosis, treatment and prevention of infectious diseases45. One approach is to use microarrays to compare the genome sequence of unsequenced strains with a reference strain, or to analyze the genomic diversity between strains with different pathogenic spectra. Salama and co-workers, for example, used a wholegenome H. pylori DNA microarray to characterize the genetic diversity of 15 clinical H. pylori isolates46. DNA microarrays are also used to determine the expression level of each gene in a genome and compare the transcriptional profiling of several conditions. Microarray analyses have been used to determine how H. pylori adapts to the low pH environment of the stomach30. Another application is to analyze variations in gene expression at different stages of infection. This information can help to characterize the natural history and pathogenesis of a bacterial infection.
Comparative genomicsComparative genomics is the study of the relationships between the genomes of different species or strains and involves the use of experimental procedures or computer programs that search for regions with similarity between genomes. Genome comparisons are likely to reveal important information about the functions and evolutionary relationships of the vast majority of genes in any genome47. Analyzing genomes from closely related species can accelerate their functional annotation, track the spread of transposons, antibiotic resistance genes, and extrachromosomal elements between species, identify potentially antigenic proteins for diagnostic purposes48, and provide insight into the adaptation of microbes to their unique ecological niches40. Comparing the genomes of pathogenic bacteria to genomes of normal human flora or non-pathogenic strains is an important way to discover the genetic mechanisms of pathogenesis40. As more genome sequences from different bacteria and strains become available, these comparisons will provide more valuable information.
Identification of virulence factors and pathogenicity islands (PAIs)One of the most promising applications of pathogenic genome analysis is the identification of virulence genes. It is important to determine the virulence genes of a pathogen to understand the pathology it causes and to develop new antimicrobial agents. Virulence genes are not usually isolated in the genomes of bacterial pathogens. Instead, several virulence factors are usually found in specific regions of the chromosomes of both gram-positive and gram-negative bacteria, forming the so-called pathogenicity islands (PAIs). These regions, which are up to 200 kb in size, often have specific insertion sequences (IS) at their ends that facilitate their translocation and insertion between microorganisms. PAIs are a subset of genomic islands (GIs). GIs and PAIs have generally been found to differ significantly in G + C content from the average genome. A number of bioinformatic tools and databases for island detection have been developed (table 2). There are several approaches for identifying GIs. One of them, known as the genome composition approach, involves searching for regions with DNA signatures (such as G + C content or dinucleotide bias) that are distinct from those of the rest of the genome49,50. An alternative to identify GIs and virulence genes is comparative genomic analysis. There are two options for this purpose26: The first is to compare closely related genomes and identify differences that may correlate with pathogenicity. The second is to compare very different genomes of species that cause similar infections and identify similarities in their genomes that may correlate with a particular phenotype.
Clinical applicationsComplete sequencing of a pathogen has many clinical applications. An important one is the development of new antimicrobial agents. To this end, it is necessary to identify targets in the genome that are essential to its survival during infection (eg, genes homologous to essential genes of similar microorganisms), test large chemical libraries of potential antimicrobials, and modify the candidate molecules to improve their efficacy and reduce toxicity41. Another application is the development of new vaccines through identification of potential antigens. This is achieved by cloning and expressing each gene of a pathogenic bacterium in a surrogate host such as E. coli and testing its immunogenicity in an animal model. This heterologous expression makes it easier to obtain and purify the potential antigenic protein and overcomes the difficulty of growing certain pathogens in the laboratory41. Other important applications that follow complete sequencing of a genome include more accurate disease diagnosis by identifying unique sequence candidates for PCR-based assays, and finding candidates for immunodiagnostic tests by heterologous expression of each coding sequence, as discussed above41. Microbial genomics can help to elucidate the mechanisms of the high antibiotic resistance that some bacteria (e.g. M. tuberculosis) naturally present. Knowledge of these mechanisms will promote better use of existing drugs and help in the development of new ones.
Complete genome analysis of a bacterium will also provide insight into the evolutionary forces involved in the adaptation of microbes to their unique ecological niche and determine which factors shape host-pathogen interaction, microbiological virulence, and host response to infection. An interesting aspect of the study of microbial genomes is to analyze how usually non-pathogenic microbes can, under certain conditions, cause infections in healthy individuals. Many of these microorganisms are members of the flora of our skin and mucous membranes. Small differences between strains of such organisms determine whether they will be able to act as opportunistic pathogens under special conditions. Detailed analysis of the differences observed when two strains of the same species of bacteria are compared shows that several small changes (eg, a cluster of single nucleotide polymorphisms) suffice to help bacteria adapt to different environments and allow them to act as opportunistic pathogens51.
Misuse of genomic knowledgeAnother hypothetical outcome of genomic knowledge of bacterial pathogens is that this data could be misused to create modified pathogens with greater virulence or resistance to antimicrobials. This would require a huge amount of knowledge about several aspects of the mechanisms of infection, as well as other biological details about the microorganism involved, but the possibility cannot be ignored. To minimize such misuse, it is imperative to obtain the genome sequence of pathogens that can be used as biological weapons52. It is generally accepted that open publication of such genomic data has more advantages than disadvantages.

La Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica agradece la colaboración de:
SOCIOS PROTECTORES
-
- Pfizer GEP, S.L.U.
- Shionogi, S.L.U.
- AbbVie Spain, S.L.U.
- Gilead Sciences, S.L.U.
- Vircell Spain, S.L.U.
- Roche Diagnostics, S.L.U.
- ASTRAZENECA FARMACÉUTICA SPAIN, S.A.
- Qiagen Iberia S.L.
- Laboratorios ViiV Healthcare, S.L.
SOCIOS PATROCINADORES
- Becton Dickinson S.A.U.
- Janssen-Cilag, S.A.
- Angelini Farmacéutica, S.A.
- Biomerieux España, S,A.U.
- Merck Sharp & Dohme de España, S.A.
- Hologic Iberia, S.L.U.
- Laboratorios Menarini S.A.
- Accelerate Diagnostics S.L.
- Abbott Laboratories, S.A.
- Cepheid Iberia S.L.U.
- Illumina Productos de España. S.L.U.
- DiaSorin Iberia, S.A.
- GlaxoSmithKline, S.A.
- Sysmex España S.L.

Enfermedades Infecciosas y Microbiología Clínica sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas