




Introducción
La tuberculosis es una enfermedad prevenible y curable. Uno de los elementos claves para su control es la identificación y tratamiento de aquellos sujetos infectados por Mycobacterium tuberculosis y con riesgo para desarrollar enfermedad tuberculosa1-3. Este aspecto resulta especialmente relevante en pacientes infectados por el virus de la inmunodeficiencia humana (VIH), en los que la probabilidad de progresión a tuberculosis es muy elevada4-8.
La eficacia de isoniazida (H) para prevenir el desarrollo de tuberculosis en pacientes infectados por el VIH ha sido suficientemente contrastada9-12, habiéndose demostrado que la administración diaria de isoniazida durante 12 meses (12H) reduce en un 83% la incidencia de tuberculosis4. Sin embargo se ha observado que estas pautas largas de tratamiento de la infección latente tuberculosa (ILT) pueden producir un considerable número de abandonos del tratamiento13. Por ello cabría esperar que pautas más cortas de tratamiento podrían disminuir las tasas de abandono del mismo.
En este contexto un estudio14 demostró que un régimen de 2 meses con rifampicina y pirazinamida (2RZ) era tan eficaz como la pauta de 12H, con tasas similares de toxicidad y mortalidad, pero con una tasa de cumplimiento significativamente mayor. Basándose en estos datos los Centers for Disease Control and Prevention (CDC) inicialmente consideraron como pautas electivas para el tratamiento de la ILT en pacientes coinfectados, los regímenes diarios de 2RZ o 12H15. Sin embargo, la descripción de casos de hepatitis graves en pacientes no infectados por el VIH que recibían 2RZ en el curso del tratamiento de la ILT y la observación en un ensayo clínico aleatorizado llevado a cabo entre pacientes no infectados por el VIH de una mayor incidencia de hepatotoxicidad en el grupo de pacientes que recibieron 2RZ en relación a los que recibieron 6H16, hizo recomendar no utilizar 2RZ en pacientes no infectados por el VIH y plantear dudas sobre su recomendación en pacientes infectados por el VIH17.
Presentamos un ensayo clínico aleatorizado, prospectivo y multicéntrico cuyo objetivo ha sido determinar el grado de cumplimentación y la seguridad de 2RZ en comparación a otras 2 pautas cortas de tratamiento de la ILT en pacientes infectados por el VIH.
Pacientes y métodos
Diseño y población
El estudio se diseñó como un ensayo clínico controlado, abierto, aleatorio y multicéntrico; que fue llevado a cabo entre el 1 de junio de 1994 y el 31 de diciembre de 1998 en 12 hospitales públicos españoles, uno de ellos ubicado en Madrid y los otros 11 en Andalucía. El estudio fue autorizado por los Comités de Ética e Investigación Clínica de todos los hospitales participantes y se obtuvo el consentimiento informado por escrito de todos los sujetos seleccionados en el mismo. La aleatorización de los pacientes a cada rama del estudio se realizó en cada centro mediante tablas de aleatorización facilitadas por el centro coordinador. Los criterios de inclusión en el estudio fueron: a) infección por el VIH confirmada por análisis inmunoenzimático (ELISA) y Western blot; b) edad comprendida entre 18 y 65 años; c) expectativas de vida superiores a 2 años, y d) reacción positiva a tuberculina.
El test de la tuberculina fue realizado en la cara volar del antebrazo izquierdo, con la técnica habitual de Mantoux, con 0,1 ml de tuberculina (2 UI de PPD RT-23). Se consideró positiva cualquier induración transversal mayor o igual a 5 mm. En los pacientes con reacción dudosa a la tuberculina (entre 1 y 4 mm) se repitió una semana después para evaluar el efecto booster. Los criterios de exclusión fueron: a) existencia de tuberculosis activa, b) antecedentes de tratamiento de tuberculosis o tratamiento de ILT previa, c) presencia de síntomas o de signos sugestivos de tuberculosis pulmonar o extrapulmonar (fiebre de origen desconocido, linfadenomegalias, etc.), d) historia de hipersensibilidad a los fármacos en estudio (isoniazida, rifampicina o pirazinamida), e) concentraciones plasmáticas de aspartato-aminotransferasa y/o de alanino-aminotransferasa iguales o superiores al cuádruple de sus valores normales, de bilirrubina total superiores 2 mg/ml, y/o de creatinina superiores a 2 mg/ml, f) gestación y g) realización simultánea de otros tratamientos incompatibles con alguno de los fármacos utilizados en el estudio.
Aleatorización e intervenciones
Los pacientes fueron aleatorizados a uno de los siguientes 3 brazos de tratamiento: isoniazida durante 6 meses (6H), rifampicina más isoniazida durante 3 meses (3HR) y 2ZR. Los fármacos fueron autoadministrados diariamente por los propios pacientes, por vía oral, en las siguientes dosis: isoniazida: 5 mg/kg/día (máxima 300 mg/día); rifampicina: 10 mg/kg/día (máxima 600 mg/día); pirazinamida: 1.500 mg/ día para los pacientes de peso inferior a 50 kg, 2.000 mg/día para los de peso comprendido entre 50 y 70 kg, y 2.500 mg/día para los de peso superior a 70 kg.
En todos los sujetos participantes se realizó un estudio basal que incluyó: historia clínica y epidemiológica, exploración física, radiografía de tórax, hemograma, concentraciones séricas de creatinina, nitrógeno ureico en sangre, ácido úrico, aspartato-aminotransferasa, alanino-aminotransferasa, fosfatasa alcalina y bilirrubina total, así como un recuento de linfocitos T CD4+.
Durante el período de tratamiento los pacientes fueron evaluados, con periodicidad quincenal en los primeros 2 meses y mensual posteriormente. En cada visita de evaluación se recogía el cumplimiento terapéutico, los posibles efectos adversos de los fármacos y se realizaba un hemograma y un estudio bioquímico de función hepática y renal.
La adherencia al tratamiento fue evaluada en cada revisión por el médico responsable mediante entrevista al paciente. Se definió el cumplimiento terapéutico como la toma de, al menos, el 80% de las dosis totales de los fármacos prescritos. Se suspendió el tratamiento cuando así lo desearon los pacientes o por cualesquiera de los siguientes motivos: aparición de efectos adversos grados III o IV atribuibles a los fármacos en estudio; aumento de las cifras de aspartato-aminotransferasa y/o de alanino-aminotransferasa al triple o más de sus valores basales; desarrollo de tuberculosis; o diagnóstico de enfermedades que hicieran aconsejable la interrupción del tratamiento.
Una vez finalizado el tratamiento de la ILT, se realizó un seguimiento clínico de los pacientes durante 2 años durante el cual se efectuó un estudio idéntico al basal cada 6 meses. En los casos sospechosos de tuberculosis se realizaron las exploraciones complementarias necesarias para llegar a un diagnóstico, incluyendo siempre tinciones de ZiehlNeelsen y cultivos en medio de Löwenstein-Jensen en los fluidos o en los especímenes de los tejidos obtenidos. Si estos últimos resultaban positivos se procedía de forma sistemática a la tipificación de la micobacteria y a la realización de pruebas de sensibilidad de la misma.
Variables de valoración y seguimiento
Las variables de valoración del estudio fueron la suspensión del tratamiento por toxicidad o por cualquier otro motivo (interacciones medicamentosas, abandono voluntario, etc.) y el desarrollo de tuberculosis activa. Se consideró como tuberculosis confirmada el aislamiento del M. tuberculosis de cualquier muestra, como tuberculosis probable la presencia de bacilos ácido alcohol resistentes en cualquier muestra sin confirmación o tipificación posterior en los cultivos, y como tuberculosis posible a la enfermedad clínicamente compatible y con respuesta al tratamiento antituberculoso.
Tamaño muestral y análisis estadístico
El estudio fue diseñado como estudio piloto con un tamaño muestral de 100 pacientes por rama de tratamiento. El análisis de variables continuas se realizó mediante la prueba de la t de Student o el test de Mann-Whitney. El análisis de las variables cualitativas se realizó mediante la prueba de la chi cuadrado (χ2) o el test de Fisher. La tasa de incidencia de tuberculosis/100 personas/año se calculó dividiendo los casos por la suma de los tiempos de seguimiento de cada sujeto. El riesgo relativo de desarrollo de tuberculosis se realizó mediante el cálculo de la razón de riesgo a través de una regresión de riesgos proporcionales o regresión de Cox.
Resultados
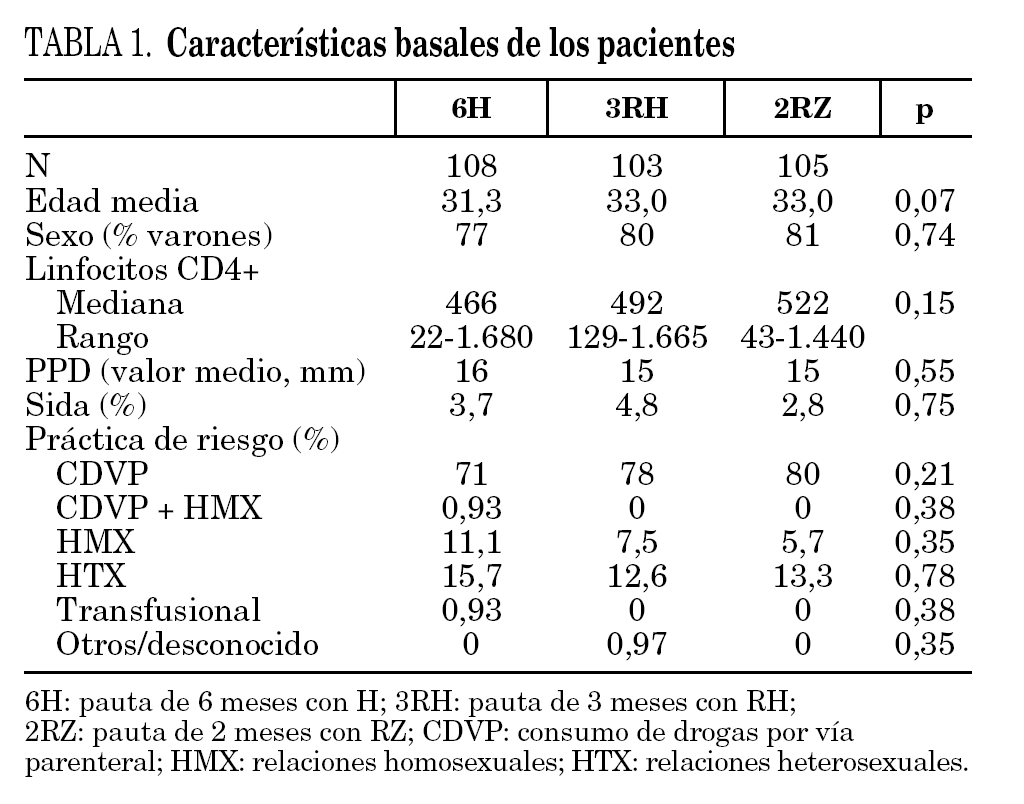
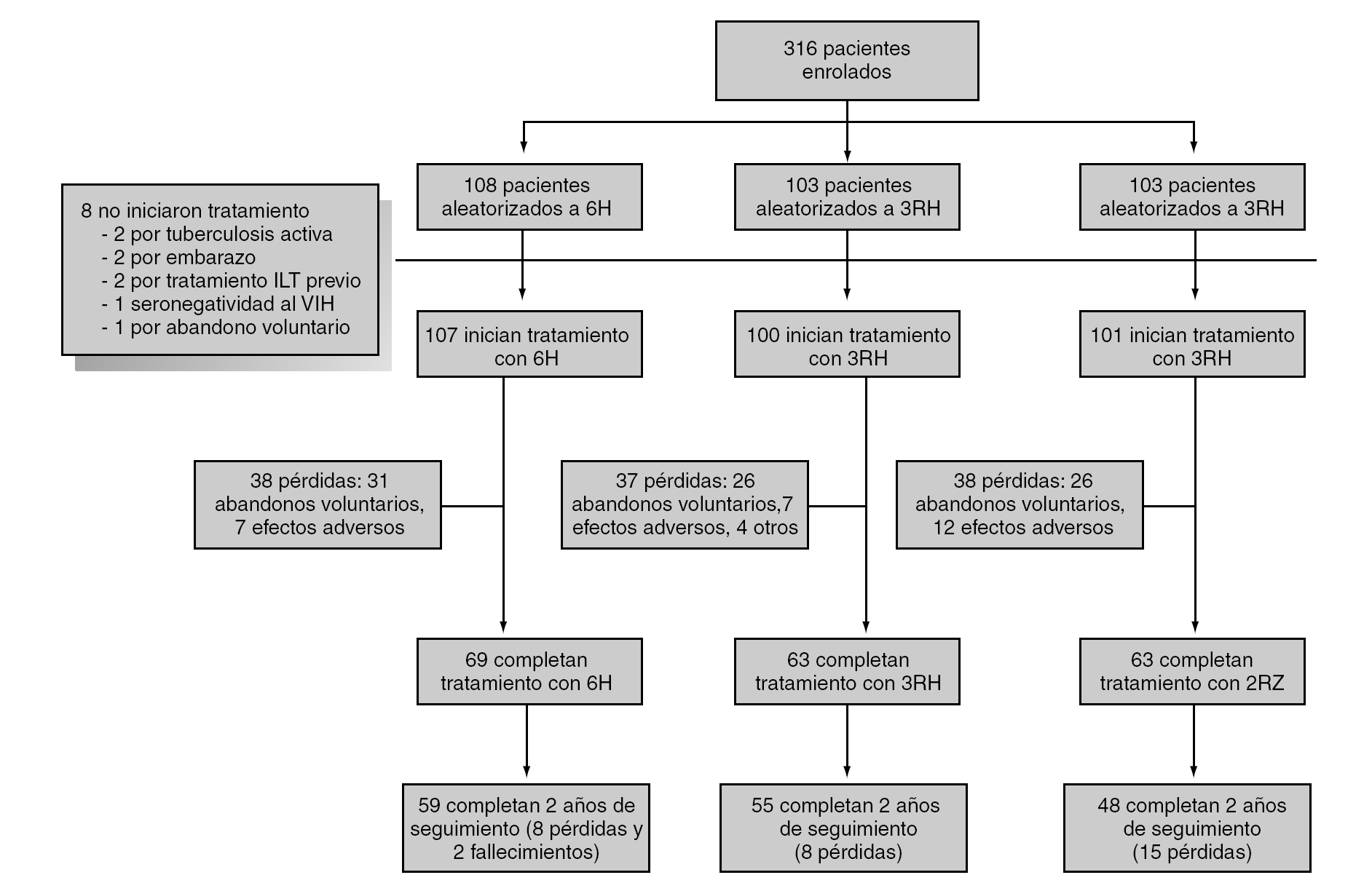
Durante el período de selección fueron incluidos en el estudio 316 pacientes cuyas características basales, equiparables en los diferentes grupos de estudio se describen en la tabla 1. Tras la aleatorización 8 pacientes no iniciaron el tratamiento (dos por diagnóstico de tuberculosis activa, dos por embarazo, dos por haberse comprobado la realización de tratamiento de la ILT previamente, uno por haberse descartado infección por el VIH y uno por abandono voluntario). No se apreciaron diferencias entre los grupos de estudio en cuanto a pérdidas durante el período de tratamiento o durante el período de seguimiento (fig. 1). Un total de 4 pacientes en tratamiento con metadona, y asignados a regímenes que contenían rifampicina, abandonaron voluntariamente el estudio para evitar los ajustes en la dosis de metadona.
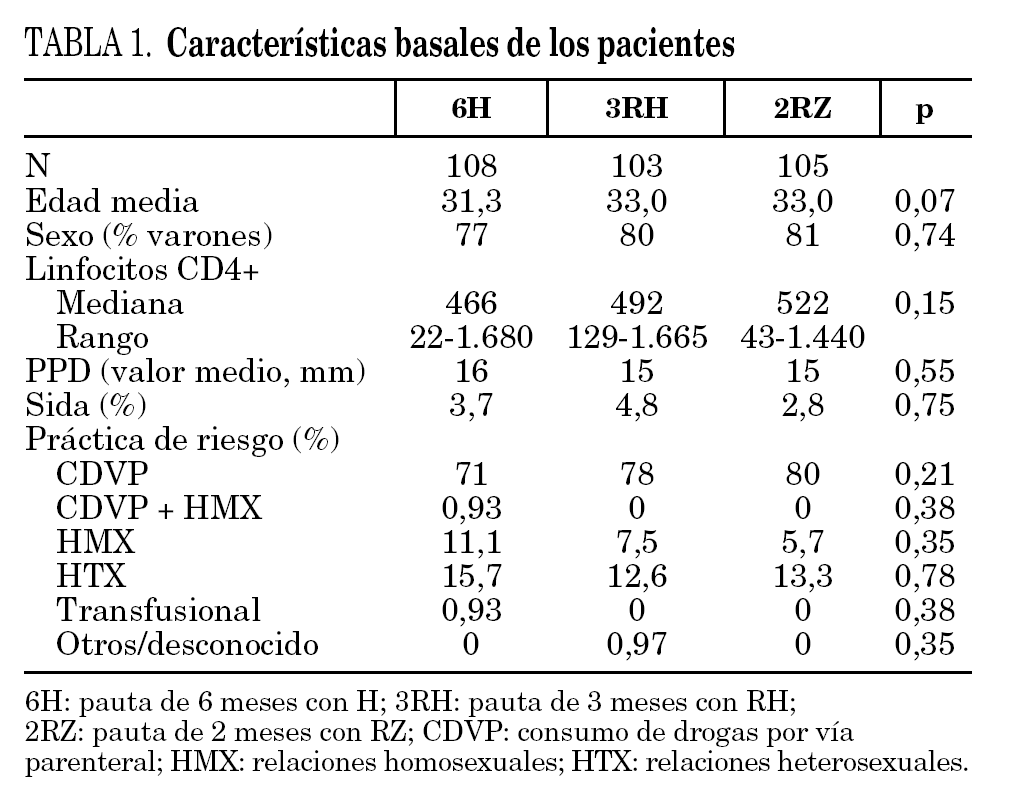
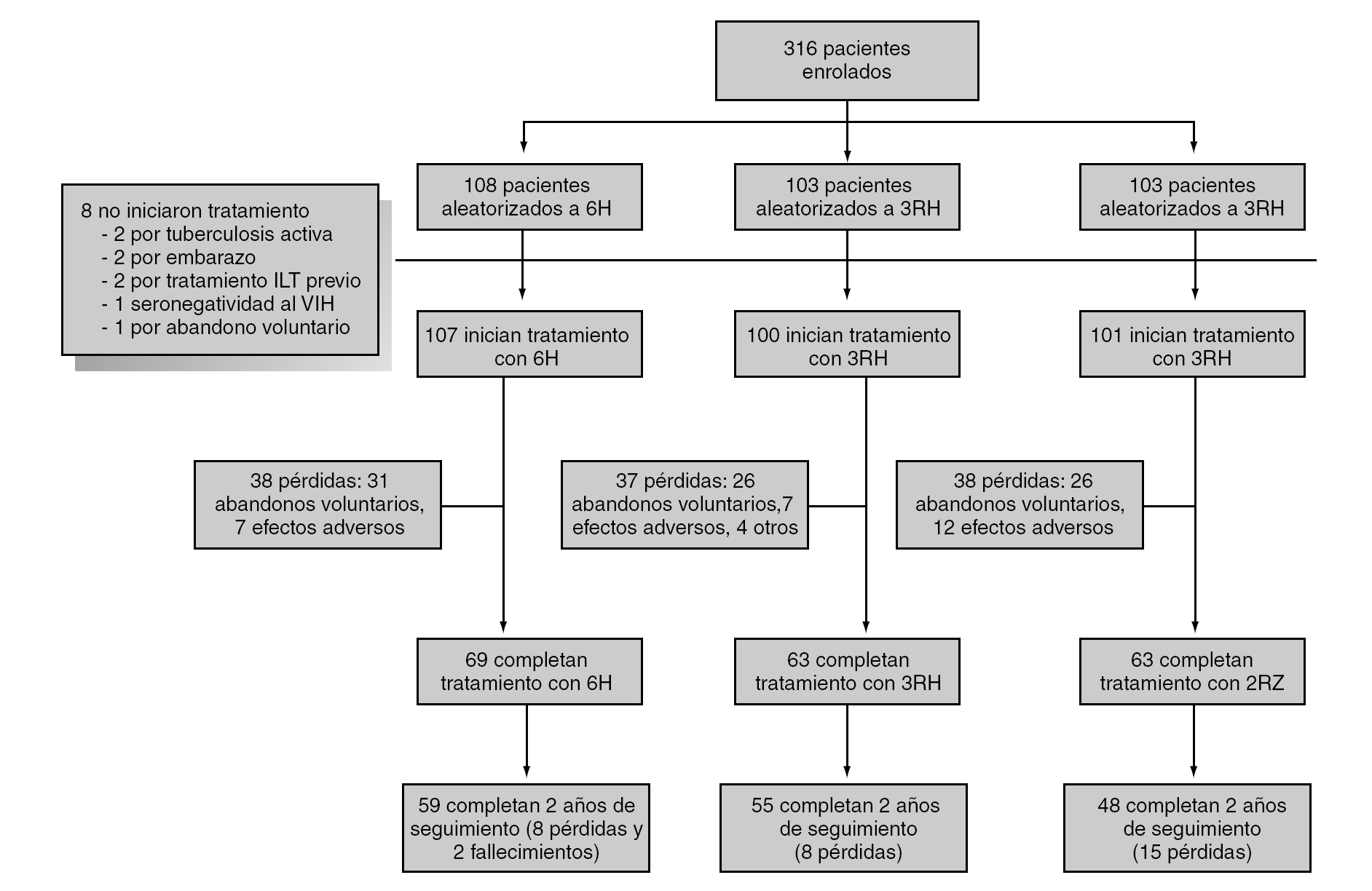
Figura 1. Desarrollo del estudio.
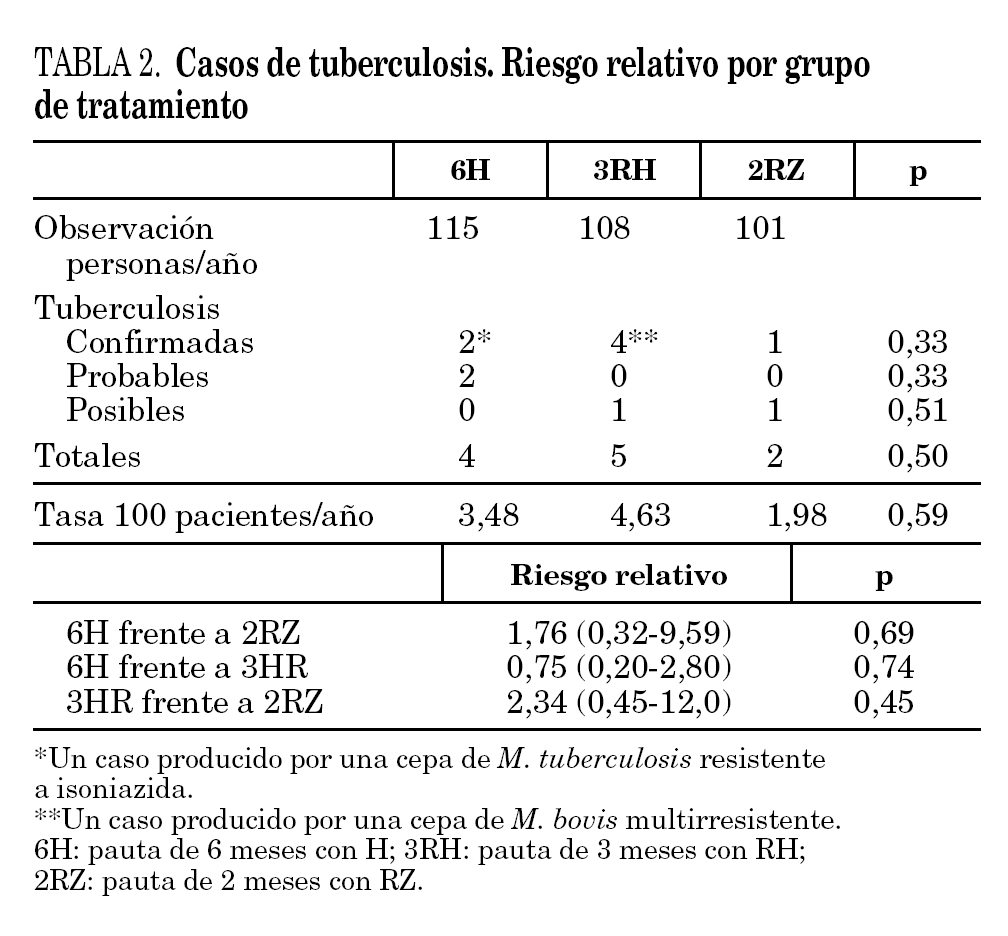
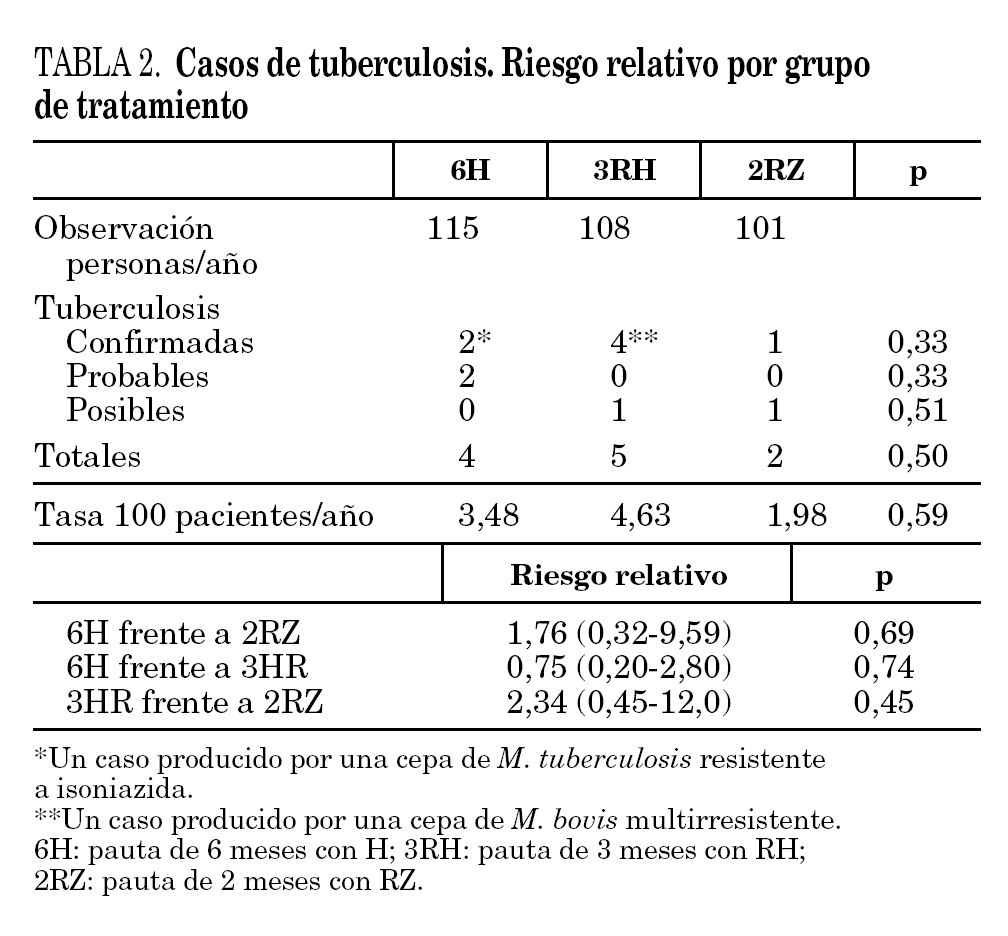
El período de observación tras la finalización del tratamiento fue de 115, 108 y 101 personas/año, respectivamente, para 6H, 3RH y 2RZ. Durante el seguimiento se produjeron 11 casos de tuberculosis (tabla 2): cuatro en el brazo 6H, cinco en el 3RH y dos en el 2RZ. En 7 casos se estableció el diagnóstico de tuberculosis confirmada, en dos de tuberculosis probable y en dos de tuberculosis posible. Los casos de tuberculosis aparecieron tras una media de 11,2 meses tras la finalización del tratamiento (rango 1-24; mediana 9 meses). Las tasas de tuberculosis en los distintos grupos fueron: 3,48 casos por 100 personas/año en el brazo 6H, 4,63 casos por 100 personas/año en el 3RH y 1,98 casos por 100 personas/año en el 2RZ. Los casos de tuberculosis y el riesgo relativo con el intervalo de confianza de los diferentes regímenes entre sí se recogen en la tabla 2.
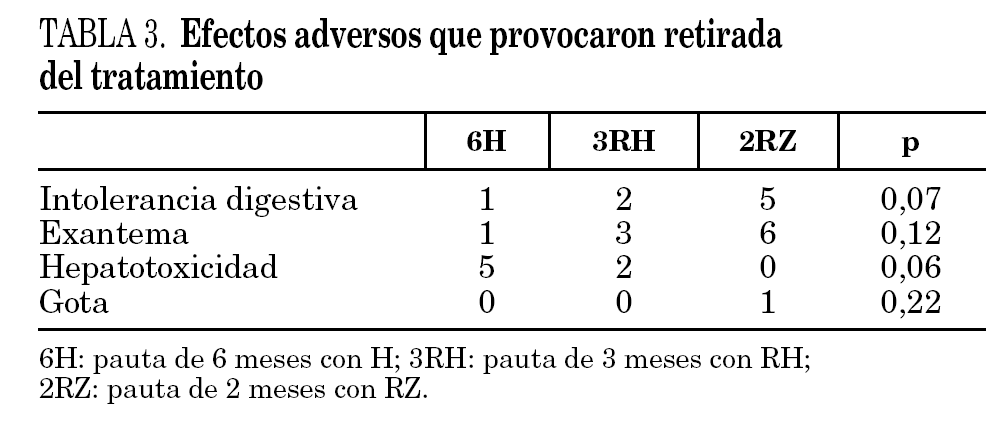
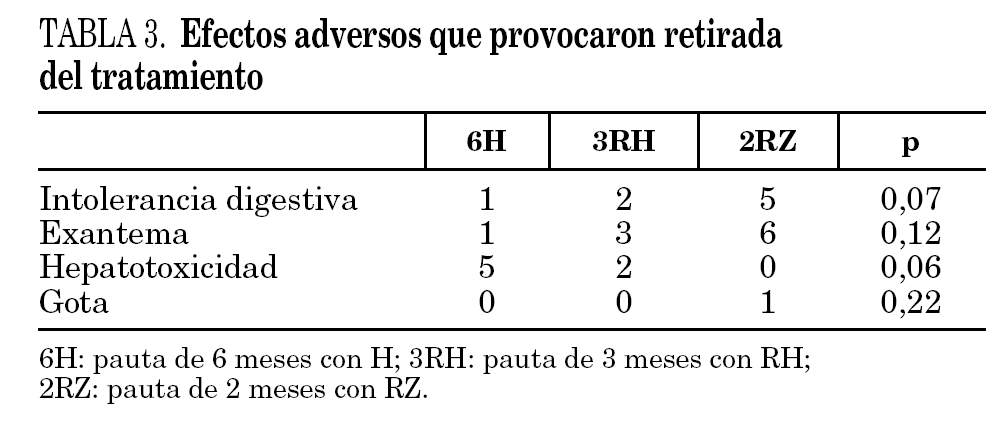
En 26 pacientes se presentaron reacciones adversas que obligaron a retirar la medicación que se pueden observar en la tabla 3. En los 7 pacientes en los que se interrumpió el tratamiento por hepatotoxicidad, la interrupción de se realizó por elevación asintomática de las transaminasas, definida como un aumento de transaminasas superior a tres veces el valor basal. En ningún caso se apreció el desarrollo de hepatitis sintomática.
Ningún paciente falleció durante el período de tratamiento. Dos pacientes fallecieron durante el seguimiento, uno por miocarditis por Toxoplasma gondii y otro por leucoencefalopatía multifocal progresiva.
Discusión
La infección por el VIH ha sido considerada como el factor de riesgo más importante para el desarrollo de tuberculosis activa en sujetos con infección latente por M. tuberculosis8. España es el país occidental con mayor tasa de coinfección VIH-M. tuberculosis18 con el agravante adicional de que la práctica principal de riesgo para el VIH es el consumo de drogas por vía parenteral (CDVP)19, y ésta, por sí misma, constituye uno de los factores de riesgo para una deficiente cumplimentación de cualquier terapia20. Con estas peculiares circunstancias, adquiere un especial interés la investigación de nuevas pautas cortas de tratamiento de la ILT que pudieran ayudar a conseguir unas mejores tasas de cumplimentación. El objetivo del estudio ha sido evaluar el grado de cumplimentación, la tolerancia y la eficacia de 3 pautas cortas de tratamiento de la ILT en pacientes coinfectados por VIH y M. tuberculosis; 6H, 3RH y 2RZ.
La primera de las pautas evaluada ha sido 2RZ, que fue considerada por los CDC como una de las 2 pautas electivas de tratamiento de la ILT en pacientes infectados por el VIH (12H y 2RZ)16, desde que Gordin et al14, confirmando los resultados obtenidos en un estudio previo18, demostraron que no sólo era tan eficaz como la pauta de 12H sino que también mejoraba la tasa de cumplimentación (80 y 69%, respectivamente). Sin embargo, entre febrero y agosto de 2001 se comunicaron a los CDC 21 casos de hepatitis graves o fatales en sujetos sin infección por el VIH que recibían 2RZ como tratamiento de ILT. Ello hizo desaconsejar su uso en pacientes no infectados por el VIH17.
Los datos disponibles de seguridad y eficacia de la pauta 2RZ en el tratamiento de la ILT en pacientes infectados por el VIH proceden de ensayos clínicos controlados y no demuestran ni sugieren un aumento del riesgo de hepatitis grave entre pacientes que reciben 2RZ21. Por el contrario, un amplio ensayo clínico demostró que los pacientes que recibían 2RZ presentaban una menor incidencia de elevación de transaminasas que los que recibían 12H14. Tampoco se demostró mayor riesgo de hepatotoxicidad en 2 ensayos clínicos en los que se administró 2RZ en régimen de 2 dosis semanales4,18, ni en un ensayo clínico controlado que en el que se evaluó la eficacia y seguridad de 2RZ en pacientes infectados por el VIH con anergia cutánea22.
En nuestro estudio tampoco se observa una mayor incidencia de hepatotoxicidad entre los pacientes que recibieron esta pauta. Ninguno de los pacientes que recibieron 2RZ interrumpieron el tratamiento por el desarrollo de hepatotoxicidad. Por lo tanto nuestro estudio, al igual que otros4,14,21,22, no confirma las sospechas de que la pauta 2RZ pueda estar relacionada con una mayor incidencia de hepatitis graves en pacientes infectados por el VIH.
Entre pacientes sin infección por el VIH el régimen 6H, aunque ha demostrado ser capaz de reducir el riesgo de reactivación tuberculosa entre 60-90%7,23, ha demostrado ser menos eficaz que 12H24. Los estudios que han evaluado la eficacia de 6H han obtenido resultados discrepantes. Mientras que Whalen et al5 demostraron que disminuía en un 68% el riesgo de desarrollar tuberculosis en pacientes infectados por el VIH con ILT, en otro estudio de similares características, este régimen no demostró ser superior al placebo7. En otros estudios en cambio se ha demostrado la eficacia de 6H administrada dos veces por semana en pacientes con ILT6,21.
La asociación 3RH, la tercera de las pautas evaluadas en este estudio, ha sido menos estudiada. Sin embargo, resulta especialmente interesante no sólo porque permitiría acortar el tiempo de tratamiento sino porque además facilitaría el uso combinado de ambos fármacos en el mismo compuesto farmacéutico lo que, probablemente, redundaría en una mejor adherencia. Este esquema proporcionó un nivel de protección similar al de 6H y permitió disminuir en un 60% el riesgo de tuberculosis frente al placebo en una amplia población de pacientes con ILT5. Otro estudio realizado en España25, comparó 3RH con 12H aunque no pudo obtener resultados definitivos por incluir a un pequeño número de sujetos en su mayoría anérgicos (sólo el 35% eran reactores a la tuberculina), de los que muy pocos llegaron a finalizar el tratamiento de la ILT.
En nuestro estudio el riesgo relativo de desarrollo de tuberculosis con las pautas de 6H y 3RH resultó 1,76 y 2,34 veces mayor que con la de 2RZ sin que dicha diferencia fuese estadísticamente significativa, aunque debido a la falta de poder estadístico del estudio no se pueden descartar diferencias clínicamente relevantes entre los grupos. Por otro lado el 8,4% de los pacientes incluidos en el estudio fueron retirados del mismo por efectos adversos sin que se apreciaran diferencias entre los 3 grupos. Estos resultados podrían sugerir que ambas pautas proporcionan un grado de protección y seguridad similar, y podrían ser útiles para prevenir la tuberculosis en estos pacientes. Sin embargo, nuestro estudio tiene diversas limitaciones que obliga a interpretar con cautela estos resultados. La principal limitación de nuestro estudio es que carece de la potencia suficiente para poder demostrar equivalencia entre las pautas evaluadas y por este motivo no permite obtener conclusiones al respecto. Por otro lado, en el diseño del estudio realizado en 1993-1994, no se tuvo en cuenta registrar del empleo de tratamiento antirretroviral de gran actividad (TARGA). Por ello el uso de TARGA durante el seguimiento no fue recogido prospectivamente y no ha sido posible evaluar su efecto en la incidencia de tuberculosis. Por último el porcentaje de abandonos del tratamiento fue elevado (27%) en cualquiera de las pautas empleadas y sin diferencia entre ellas.
Aunque diversos estudios han demostrado que la utilización de pautas cortas del tratamiento de la ILT puede incrementar su cumplimentación14,26-28, también es conocido que el CDVP es factor de riesgo para la incorrecta adherencia a cualquier tratamiento11,17. El alto porcentaje de abandonos observado en nuestro estudio pudiera estar en relación con la elevada proporción de pacientes CDVP incluidos en el mismo. Este hecho ha sido observado también en otros estudios realizados en nuestro país25. El hecho de que incluso en las condiciones casi ideales de un ensayo clínico y utilizando pautas cortas del tratamiento de la ILT el porcentaje de pacientes que la finalizan sea bajo debería ser motivo de reflexión. Explorar nuevas estrategias que mejoren la cumplimentación del tratamiento de la ILT en sujetos con alto riesgo de abandono de la misma debe ser un objetivo prioritario para el control de la tuberculosis29.
Por último alguno de los casos de tuberculosis detectados durante el seguimiento pudieran ser no atribuibles al fracaso de la pauta de tratamiento. En pacientes infectados por el VIH se ha descrito el desarrollo de tuberculosis por la rápida progresión de una reinfección reciente30. Éste podría ser el probable mecanismo de la enfermedad en algún paciente, especialmente en un paciente que presentó tuberculosis producida por una cepa de Mycobacterium bovis multirresistente en el curso de un brote de tuberculosis multirresistente31.
In memoriam
Al Dr. Rafael Rey Durán amigo, maestro e impulsor del proyecto, en el que trabajó como investigador principal y que falleció antes de que éste viera la luz.
Agradecimientos
Al Dr. Agustín Gómez de la Cámara de la Unidad de Investigación del Hospital 12 de Octubre de Madrid por su ayuda en las valoraciones estadísticas.
Conflicto de intereses
No existe conflictos de intereses en ninguno de los autores del manuscrito.
Número de registro del Ensayo clínico: NCT0040254 (Nacional Institutes of Health, Food and Drug Administration, USA).
Estudio financiado por el Fondo de Investigaciones Sanitarias (expediente n.º: 94/0071).
Correspondencia:
Dr. A. Rivero.
Sección de Enfermedades Infecciosas. Hospital Universitario Reina Sofía.
Avda. Menéndez Pidal, s/n. 14004 Córdoba. España.
Correo electrónico: ariveror@saludalia.com
Manuscrito recibido el 1-6-2006; aceptado el 16-12-2006.

