





Introducción
Los ácidos nucleicos y las proteínas, macromoléculas comunes a todos los seres vivos, cambian con el tiempo. Por ello, pueden considerarse como cronómetros moleculares o documentos de la historia evolutiva. Asumiendo que los cambios se producen al azar y que aumentan con el tiempo de manera lineal, las diferencias en la secuencia de los monómeros (nucleótidos o aminoácidos) que integran macromoléculas homólogas, presentes en dos formas de vida, reflejan la distancia evolutiva existente entre ellas. Esta idea, introducida por Zuckerkandl y Pauling1, se ha venido utilizando durante décadas para establecer las relaciones filogenéticas entre los seres vivos, creando un marco apropiado para su clasificación e identificación.
El ARN ribosómico (ARNr) 16S es la macromolécula más ampliamente utilizada en estudios de filogenia y taxonomía bacterianas. Su aplicación como cronómetro molecular fue propuesta por Carl Woese (Universidad de Illinois) a principios de la década de 19702. Los estudios de Woese originaron la división de los procariotas en dos grupos o reinos: Eubacteria y Archaeobacteria, cuya divergencia es tan profunda como la encontrada entre ellos y los eucariotas. Además, permitieron establecer las divisiones mayoritarias y subdivisiones dentro de ambos reinos3. Posteriormente, Woese introdujo el término dominio para sustituir al reino como categoría taxonómica de rango superior, y distribuyó a los organismos celulares en tres dominios: Bacteria, Archaea y Eukarya, el último de los cuales engloba a todos los seres eucariotas4. Desde entonces, el análisis de los ARNr 16S se ha utilizando ampliamente para establecer las relaciones filogenéticas dentro del mundo procariota, causando un profundo impacto en nuestra visión de la evolución y, como consecuencia, en la clasificación e identificación bacteriana. De hecho, las ediciones vigentes de los dos tratados fundamentales de bacteriología, el Bergey's Manual of Systematic Bacteriology (http://www.springer-ny.com/bergeysoutline/ main.htm) y The Prokaryotes (http://www.prokaryotes.com), basan su estructuración del mundo procariota en las relaciones filogenéticas establecidas con esta macromolécula.
Los ARNr 16S pueden caracterizarse en términos de secuencia parcial, mediante el método de catalogación de oligonucleótidos, utilizado en los estudios pioneros de Woese. Siguiendo esta técnica, el ARNr 16S marcado in vivo, y purificado, se trata con la enzima ribonucleasa T1. Los fragmentos generados se separan, determinándose posteriormente la secuencia de todos aquellos que incluyan al menos seis nucleótidos (nt). A continuación, las secuencias de la colección de fragmentos correspondientes a diferentes bacterias se alinean y comparan, utilizando programas informáticos, para calcular finalmente los coeficientes de asociación. Como se verá más adelante, la secuenciación del gen que codifica el ARNr 16S ha sustituido en la actualidad a la secuenciación de catálogos de oligonucleótidos.
En microbiología clínica, la identificación rápida y correcta de los agentes patógenos es un requisito esencial para el diagnóstico y la aplicación posterior de un tratamiento adecuado. En la actualidad, la mayor parte de las bacterias de interés clínico pueden identificarse fácilmente mediante técnicas microbiológicas convencionales, que requieren el aislado previo del agente patógeno y se basan en características fenotípicas. Sin embargo, existen situaciones en las cuales la identificación fenotípica necesita mucho tiempo, resulta difícil o, incluso, imposible. En estas circunstancias, la identificación molecular basada en el análisis del ARNr 16S (o del gen que lo codifica) puede representar una ventaja tanto en tiempo como en precisión, llegando incluso a competir de manera favorable con otras técnicas rápidas y eficaces, como las inmunológicas. En este artículo, revisaremos el fundamento y los aspectos técnicos de la metodología implicada, analizaremos sus ventajas e inconvenientes con respecto a métodos tradicionales, y presentaremos ejemplos de distintas aplicaciones en el campo de la microbiología clínica.
Los ribosomas, los operones ribosómicos y el ARNr 16S
Ribosomas
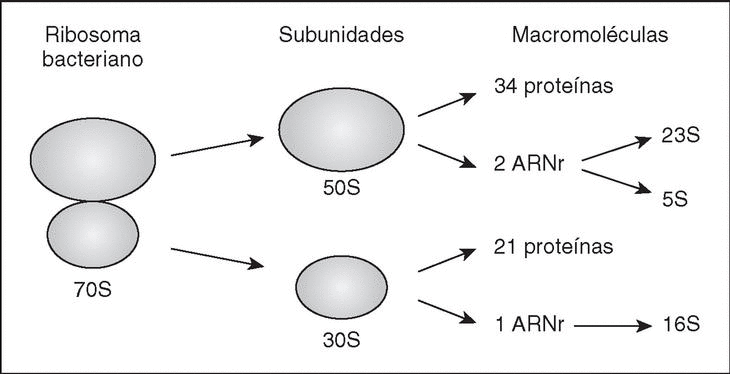
Son orgánulos complejos, altamente especializados, que utilizan los organismos para el complicado proceso de síntesis de proteínas. El ribosoma bacteriano (fig. 1) tiene un coeficiente de sedimentación de 70S (expresado en unidades Svedberg), y puede disociarse en dos subunidades, la subunidad grande (50S) y la subunidad pequeña (30S). Cada subunidad es un complejo ribonucleoproteico constituido por proteínas ribosómicas y moléculas de ARNr específicas. La subunidad 30S contiene el ARNr 16S y 21 proteínas diferentes (numeradas desde S1-S21, donde S procede de small), mientras que la subunidad 50S contiene los ARNr 5S y 23S junto con unas 34 proteínas (L1-L34; L, large).
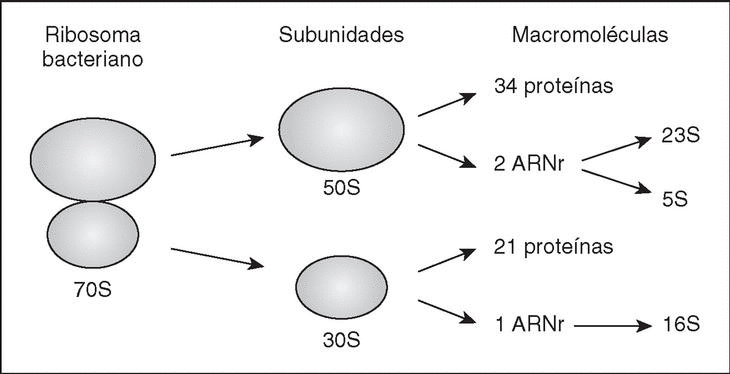
Figura 1. El ribosoma bacteriano. Se muestra un esquema de su estructura, y se indican las subunidades y macromoléculas que lo componen.
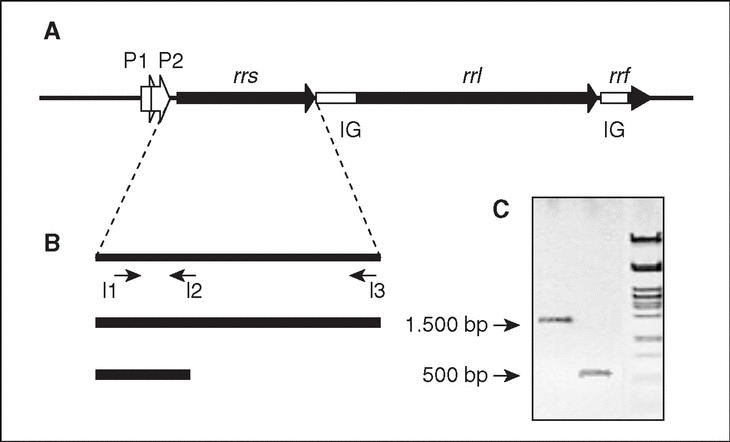
En bacterias, los genes que codifican los ARN ribosomales están organizados en operones (conjunto de genes que se transcriben a partir de la misma región promotora). Cada operón ribosómico (rrn; fig. 2) incluye genes para los ARNr 23S (rrl), 16S (rrs) y 5S (rrf), separados por regiones espaciadoras o intergénicas (IG), y contiene además genes para uno o más ARN de transferencia (ARNt). El producto de la transcripción del operón a partir de dos promotores, P1 y P2, situados en la región anterior a rrs, será procesado por el enzima ARNasa III mediante cortes en sitios específicos que separan las tres clases de ARNr, el/los ARNt y las dos IG.
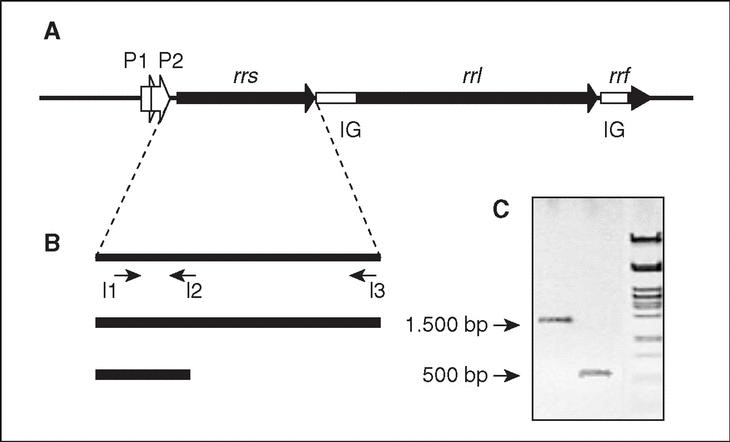
Figura 2. El operón ribosómico (rrn). A) Representación esquemática del operón, donde se muestran los genes estructurales de los tres tipos de ARNr (rrs, rrl y rrf), los promotores P1 y P2, y las regiones intergénicas (IG). B) Estrategia de amplificación del gen rrs (ADNr 16S). Se indica la posición de los iniciadores I1 (directo), I2 e I3 (reversos) utilizados para la amplificación (y posterior secuenciación) del gen completo (I1-I3; amplicón de 1500 pb, aproximadamente) o de un fragmento menor (11-I2; 500 pb correspondientes al extremo 59 del gen; ver explicación en el texto). C) Visualización de ambos fragmentos por electroforesis en gel de agarosa.
ARNr 16S
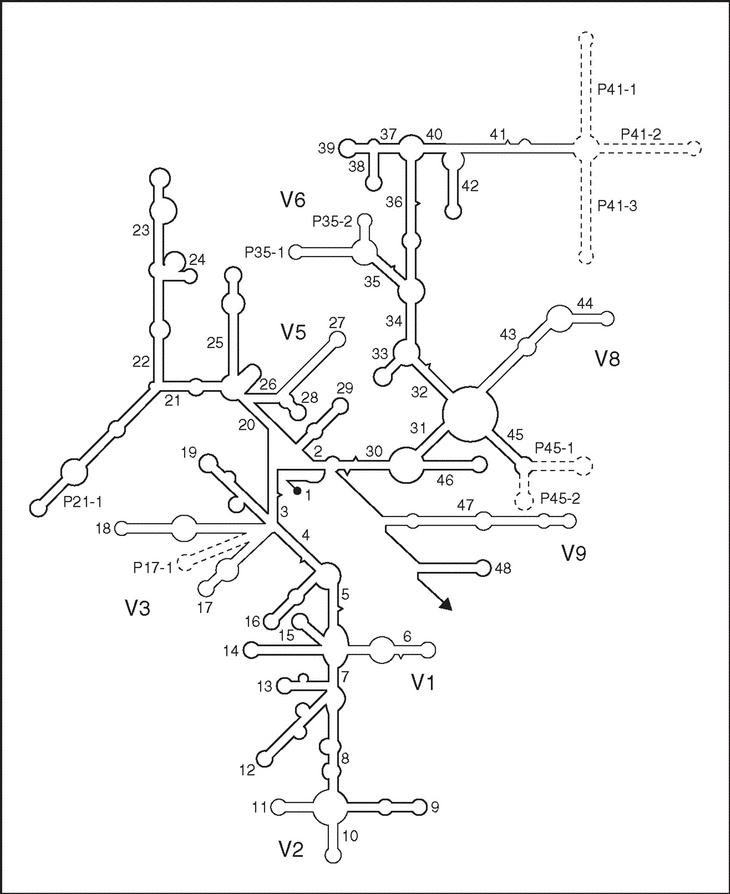
Es un polirribonucleótido de aproximadamente 1.500 nt, codificado por el gen rrs, también denominado ADN ribosomal 16S (ADNr 16S), a partir de cuya secuencia se puede obtener información filogenética y taxonómica. Como cualquier secuencia de nucleótidos de cadena sencilla, el ARNr 16S se pliega en una estructura secundaria, caracterizada por la presencia de segmentos de doble cadena, alternando con regiones de cadena sencilla5 (fig. 3). En eucariotas el ARNr 18S es la macromolécula equivalente. Dado que los ARNr 16S y 18S proceden de las subunidades pequeñas de los ribosomas, el acrónimo ARNr SSU (del inglés, small subunit) se utiliza para ambos. Los ARNr SSU se encuentran altamente conservados, presentando regiones comunes a todos los organismos, pero contienen además variaciones que se concentran en zonas específicas (fig. 3). El análisis de la secuencia de los ARNr 16S de distintos grupos filogenéticos reveló un hecho adicional de gran importancia práctica: la presencia de una o más secuencias características que se denominan oligonucleótidos firma6. Se trata de secuencias específicas cortas que aparecen en todos (o en la mayor parte de) los miembros de un determinado grupo filogenético, y nunca (o sólo raramente) están presentes en otros grupos, incluidos los más próximos. Por ello, los oligonucleótios firma pueden utilizarse para ubicar a cada bacteria dentro de su propio grupo.
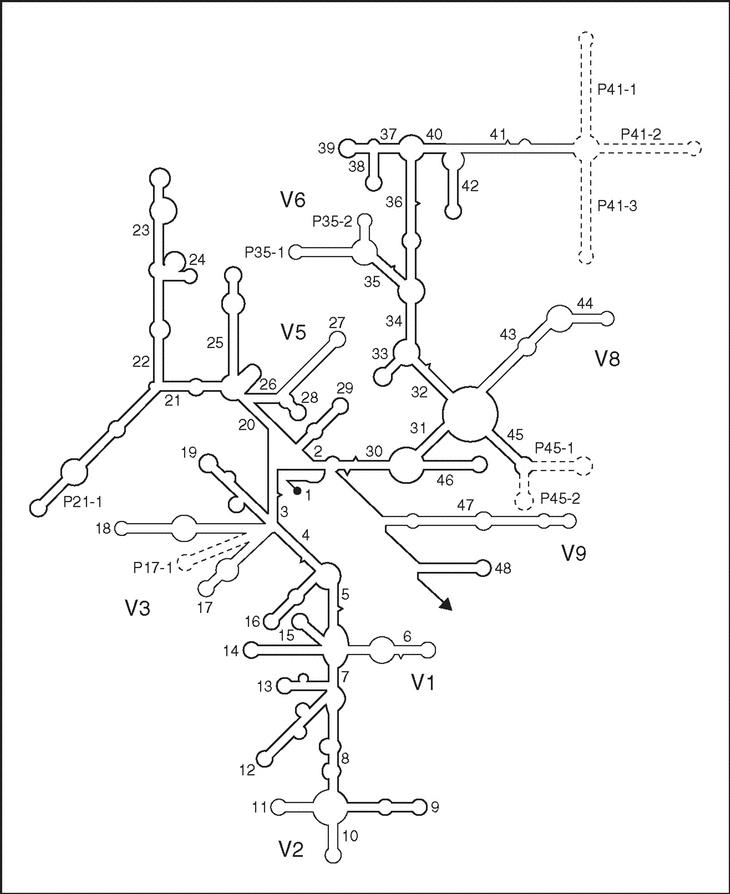
Figura 3. Estructura secundaria del ARNr 16S (tomada de Neefs et al5, con permiso de Oxford University Press; Reino Unido). Las hélices comunes a todos los seres vivos, denominadas hélices universales, se numeran de la 1 a la 48, en orden de aparición a partir del extremo 59. Las hélices específicas de procariotas se indican con Pa-b, donde a es el número de la hélice universal precedente y b el número de serie. Las regiones relativamente conservadas se presentan en negrilla. Las regiones variables, en líneas finas, se designan V1-V9, teniendo en cuenta que V4 es exclusiva de eucariotas. Las regiones que se muestran en líneas discontinuas sólo están presentes en un número limitado de estructuras.
El número de copias del operón ribosómico por genoma bacteriano varía considerablemente, de 1 a 15, siendo relativamente constante a nivel de especie, género e incluso familia7 (tabla 1). Entre las copias de los ARNr 16S codificadas por un mismo genoma se ha detectado un cierto grado de heterogeneidad (denominada microheterogeneidad). El análisis de 14 genomas bacterianos, cuya secuencia completa se encuentra disponible, indicó una divergencia máxima del 1,23%, que corresponde a los ADNr 16S de Escherichia coli (tabla 1). Además, diferentes autores encontraron variabilidad intragenómica entre los ADNr 16S de otras bacterias de interés clínico, cuyo genoma aún no ha sido secuenciado. Destaca la elevada heterogeneidad (1,43%) detectada entre las 4 copias del ADNr 16S presentes en la cepa clínica ADV 360.1 de Veillonella8. Obviamente, la variabilidad intragenómica, tiene importantes implicaciones prácticas para la identificación. Sin embargo, en la mayor parte de los casos, todas las copias del ADNr 16S de un organismos son idénticas o casi idénticas.

Características relevantes del ARNr 16S para su utilización como herramienta filogenética y taxonómica
Aunque existen cronómetros moleculares alternativos al ARNr 16S, hasta el momento ninguno ha conseguido desplazarle. De hecho, esta macromolécula presenta una serie de características, en base a las cuales fue considerado por Woese3 como cronómetro molecular definitivo:
1. Se trata de una molécula muy antigua, presente en todas las bacterias actuales. Constituye, por tanto, una diana universal para su identificación.
2. Su estructura y función han permanecido constantes durante un tiempo muy prolongado, de modo que las alteraciones en la secuencia reflejan probablemente cambios aleatorios.
3. Los cambios ocurren de manera suficientemente lenta, como para aportar información acerca de todos los procariotas y, junto con las variaciones en los ARNr 18S, a lo largo de toda la escala evolutiva. Los ARNr SSU contienen, sin embargo, suficiente variabilidad para diferenciar no sólo los organismos más alejados, sino también los más próximos.
4. El tamaño relativamente largo de los ARNr 16S (1.500 nt) minimiza las fluctuaciones estadísticas.
5. La conservación en estructura secundaria puede servir de ayuda en las comparaciones, aportando una base para el alineamiento preciso.
6. Dado que resulta relativamente fácil secuenciar los ADNr 16S existen bases de datos amplias, en continuo crecimiento.
Una vez determinada la secuencia de nucleótidos y establecidas las comparaciones, será el grado de similitud entre las secuencias de los ADNr 16S de dos bacterias lo que indique su relación evolutiva. Además, el análisis comparativo de secuencias permite construir árboles filogenéticos, que reflejan gráficamente la genealogía molecular de la bacteria, mostrando su posición evolutiva en el contexto de los organismos comparados.
Hay que tener en cuenta, no obstante, que es la comparación de genomas completos, y no la comparación de los ADNr 16S, la que aporta una indicación exacta de las relaciones evolutivas. En su ausencia, la especie bacteriana se define, en taxonomía, como el conjunto de cepas que comparten una similitud del 70% o más, en experimentos de reasociación ADN-ADN. Stackebrandt y Goebel9 demostraron que cepas con este nivel de relación presentan típicamente una identidad del 97% o más entre sus genes ARNr 16S. Así, cepas con menos del 97% de identidad en las secuencias de sus ADNr 16S es improbable que lleguen a estar relacionadas a nivel de especie. Sin embargo, existen cepas que comparten una similitud inferior al 50% en experimentos de reasociación, y son por tanto clasificadas en especies diferentes, pero presentan una identidad del 99-100% a nivel de ADNr 16S. Por ello, en taxonomía, actualmente se recomienda la identificación polifásica, que utiliza criterios fenotípicos junto con datos de secuenciación10. En microbiología clínica, esto implica que la secuenciación del ADNr 16S no aportará siempre una identificación definitiva a nivel de especie11.
Aspectos metodológicos de la identificación bacteriana mediante secuenciación del ADNr 16S
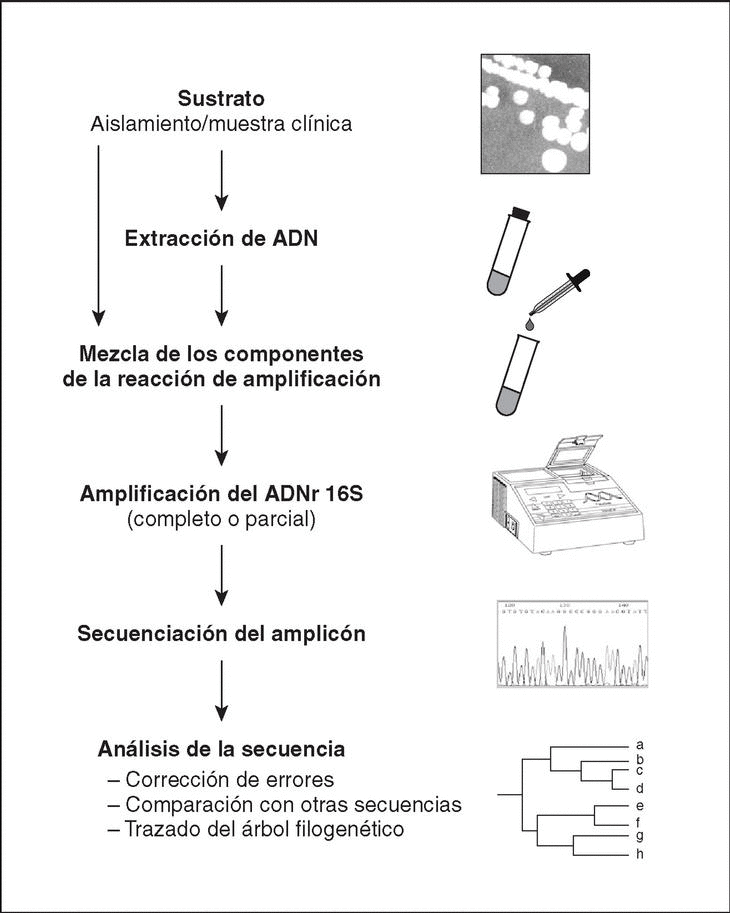
El método molecular de identificación bacteriana mediante secuenciación del ADNr 16S incluye tres etapas: a) amplificación del gen a partir de la muestra apropiada; b) determinación de la secuencia de nucleótidos del amplicón, y c) análisis de la secuencia (fig. 4).
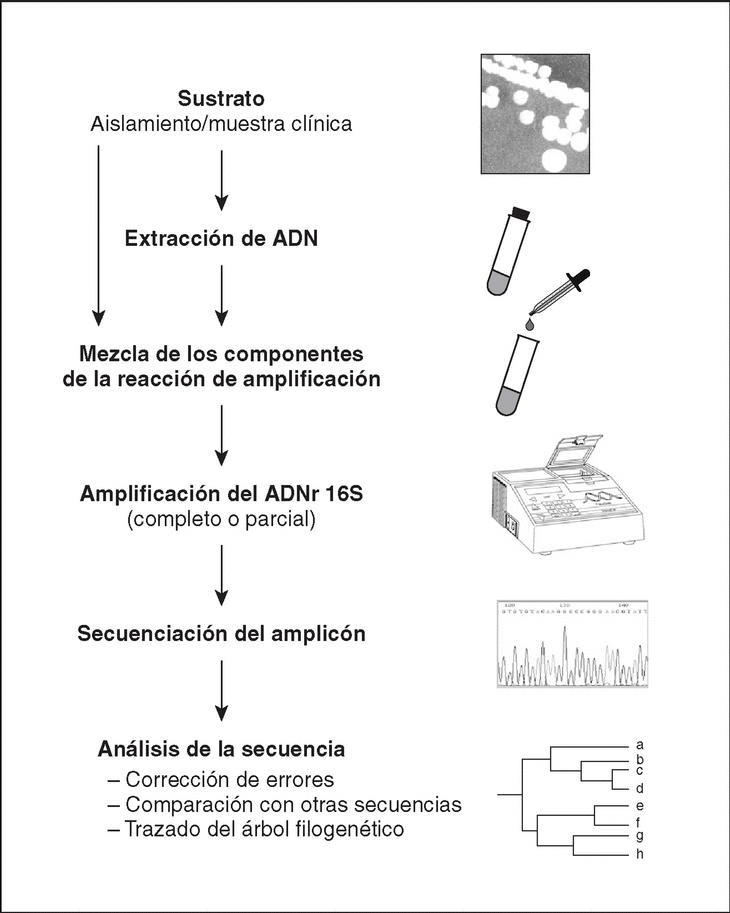
Figura 4. Etapas a seguir en el proceso de identificación bacteriana mediante secuenciación del ADNr 16S.
Amplificación
La amplificación del ADNr 16S se consigue en un termociclador, gracias a la reacción en cadena de la polimerasa (PCR). Como sustrato se utiliza normalmente ADN purificado a partir de un cultivo puro del agente patógeno. Alternativamente, el ADN podrá obtenerse directamente de la muestra clínica, en el caso de bacterias fastidiosas o no cultivables, cuando aquella proceda de órganos o tejidos normalmente estériles. Para la extracción del ADN bacteriano existen protocolos generales12, pero pueden requerirse modificaciones, dependiendo de la bacteria. Además, la amplificación también puede conseguirse directamente a partir de una colonia aislada o un cultivo líquido de la bacteria de interés, o incluso a partir de una muestra clínica, simplificando significativamente la identificación, al evitar el laborioso proceso de extracción del ADN.
Oligonucleótidos iniciadores
Para la amplificación del ADNr 16S, las regiones conservadas facilitan el diseño de oligonucleótidos iniciadores (20 nt, aproximadamente). Cuando se pretende amplificar el ADNr 16S prácticamente completo, se utilizan iniciadores diseñados en base a secuencias conservadas próximas a los extremos 59 y 39 del gen, que originan amplicones de 1.500 pb, aproximadamente. Se ha demostrado, sin embargo, que una identificación precisa no siempre requiere la amplificación, y posterior secuenciación, del ADNr 16S completo. En estas circunstancias se utilizarán oligonucleótidos que permitan la amplificación de fragmentos de menor tamaño, preferentemente las 500 pb correspondientes al extremo 59 del gen (v. más adelante). En cualquier caso, antes de pasar a la siguiente etapa es conveniente comprobar el producto mediante electroforesis en gel de agarosa, para asegurar la presencia de un único fragmento, amplicón, del tamaño adecuado (fig. 2).
Secuenciación del amplicón
En esta etapa se llevan a cabo las reacciones de secuenciación y el análisis de los productos por electroforesis. Actualmente, se emplea la secuenciación cíclica, en una reacción similar a la de amplificación, que utiliza un único iniciador por reacción y terminadores marcados con fluorocromos adecuados, que interrumpirán la síntesis de manera aleatoria, y facilitarán la detección posterior de los fragmentos interrumpidos.
La disponibilidad de secuenciadores automáticos facilita enormemente la etapa de detección. El número de bases generadas por un secuenciador automático es de 500 a 900, dependiendo del capilar utilizado en la electroforesis. Por ello, para la secuenciación de las dos cadenas del ADNr 16S completo, serán necesarios de 8 a 4 iniciadores, dos de los cuales podrán ser los mismos utilizados en la amplificación. Sin embargo, la secuencia obtenida podrá contener errores y/o presentar posiciones ambiguas (indicadas por N). Por ello, la obtención de la secuencia definitiva requiere la evaluación de los electroferogramas y la alineación de la cadena directa con la reversa, para resolver las posibles discrepancias. Así, aunque la secuenciación de una cadena del amplicón puede conducir a una correcta identificación, la calidad de la secuencia será óptima cuando la comparación de ambas cadenas se utiliza para la corrección de errores.
Ya se ha indicado que la identificación de una bacteria a nivel de especie no requiere necesariamente la secuenciación del ADNr 16S completo. De hecho, aunque existen posiciones filogenéticamente informativas a lo largo de todo el gen, la mayor variabilidad se concentra en las primeras 500 bases, correspondientes al extremo 59. Generalmente, esta secuencia de 500 bases será suficiente para la correcta identificación de un aislado clínico, necesitándose únicamente 2 iniciadores11,13-15. En cualquier caso, la secuenciación de las dos cadenas del ADNr 16S completo será necesaria a la hora de identificar nuevos patógenos.
Análisis de la secuencia
La última etapa será la comparación de la secuencia del ADNr 16S con las depositadas en bases de datos. Actualmente existen distintas bases de datos, algunas públicas, cuyo acceso es libre a través de internet, como GenBank NCBI (National Center for Biotechnology Information), EMBL (European Molecular Biology Laboratory), RDP (Ribosomal Database Project), RIDOM (Ribosomal Differentiation of Medical Microorganisms), y otras privadas, como MicroSeq (Applied Biosystems) y SmartGene IDNS (Integrated Database Network System).
RDP (http://rdp.cme.msu.edu/html/) es la principal base de datos de secuencias de ADNr (no sólo 16S, sino también 23S de procariotas, y 18S y 28S de eucariotas). Permite la comparación de secuencias on line, y ofrece otras muchas posibilidades (incluida la construcción de árboles filogenéticos; ver más adelante). En octubre de 2003 contenía más de 78.000 secuencias de ADNr 16S, que son alineadas, teniendo en cuenta la estructura secundaria de la molécula. La base de datos RIDOM (http://www.ridom.de) se limita también a secuencias de ADNr, centrándose exclusivamente en microorganismos patógenos. Como se verá posteriormente, la elección de la base de datos es importante, siendo recomendable la utilización de más de una de ellas, para comprobar si conducen al mismo resultado. Finalmente, se podrá construir un árbol filogenético, que refleja, de forma esquemática, el grado de parentesco genético entre las bacterias comparadas.
Sistemas comerciales
La identificación bacteriana basada en el análisis de la secuencia del ADNr 16S se facilita grandemente por la disponibilidad de sistemas comerciales. Entre ellos destaca MicroSeq500 (Applied Biosystems; Foster City, EE.UU.), diseñado para la identificación rápida y correcta de bacterias patógenas. Este sistema utiliza un par de oligonucleótidos que permiten amplificar un fragmento de 527-bp correspondiente al extremo 59 del ADNr 16S. En realidad se trata de una versión simplificada del "MicroSeq 16S rRNA" original (también disponible en Applied Biosystems), que sólo requiere dos iniciadores para la secuenciación de ambas cadenas, reduciendo de manera considerable el coste y el trabajo requeridos para la identificación. Como parte del sistema, se incluye una base de datos para determinar el género y la especie del aislado. Además, el software permite exportar las secuencias que podrán ser comparadas con otras bases de datos, e incluye herramientas para el análisis filogenético. La utilidad de MicroSeq500 ha sido ampliamente demostrada para una gran variedad de patógenos bacterianos (v. más adelante). El coste de cada identificación estimado en diferentes laboratorios varía considerablemente: 84 a 144 114,11, sin el gasto previo destinado a tener en cuenta equipamiento (termociclador, secuenciador automático, etc.). Sí se incluyen los gastos de extracción, material desechable, reactivos, utilización de la base de datos y salarios del personal de laboratorio. Dado que la mayor proporción corresponde a este último concepto, el coste por identificación se reduce de manera considerable al identificar más de un aislado simultáneamente.
Actualmente, otro aspecto a favor es la disponibilidad de kits comerciales independientes para cada etapa del proceso de identificación (extracción de ADN, amplificación por PCR y secuenciación), así como la posibilidad de recurrir a servicios comunes de secuenciación automática. Esto último resulta interesante, ya que sólo requiere generar el producto de PCR, cuya secuencia será determinada por el servicio de secuenciación y remitida directamente al laboratorio solicitante, por correo electrónico. En nuestro laboratorio se han utilizado, con excelentes resultados, los Servicios de Secuenciación Automática de la Universidad de Oviedo, del Hospital Central de Asturias y del Centro Superior de Investigaciones Biológicas (Consejo Superior de Investigaciones Científicas). En estos casos, partiendo del amplicón, la secuencia puede estar disponible en 24-48 h, a un precio de 10-12 1 por reacción. Muchos otros hospitales españoles disponen también de secuenciadores automáticos de ADN.
Aplicaciones de la secuenciación del ARNr 16S en el diagnóstico microbiológico
La identificación basada en la secuenciación del gen que codifica el ARNr 16S en los laboratorios clínicos se centra principalmente en cepas cuya identificación por métodos fenotípicos resulta imposible, difícil o requiere mucho tiempo, incluyendo los siguientes casos:
1. Bacterias no cultivables presentes en muestras clínicas, hecho que en ocasiones ha conducido al descubrimiento de nuevos agentes patógenos.
2. Bacterias cuyas características bioquímicas no se adaptan a las de ningún género o especie reconocido. Esta situación puede presentarse cuando se trata de patógenos nuevos, patógenos infrecuentes o también cepas de especies comunes que exhiben un perfil bioquímico ambiguo.
3. Bacterias para las cuales la caracterización fenotípica sea sustancialmente deficiente.
4. Bacterias fastidiosas, a consecuencia de sus requerimientos nutricionales.
5. Bacterias de crecimiento lento, que retrasa considerablemente la identificación convencional.
A continuación se presentan algunos ejemplos relevantes.
Entre mediados y finales de la década de 1980, pacientes infectados por el virus de la inmunodeficiencia humana (VIH), manifestaron una forma peculiar de angiomatosis, con lesiones en la piel que recordaban a las características del sarcoma de Kaposi. Estas lesiones contenían invariablemente grupos de bacilos pleomórficos, y por ello se denominó angiomatosis bacilar16. Sin embargo, a pesar de repetidos intentos, el microorganismo no logró ser cultivado. Por tanto, el análisis fenotípico, aparte de la simple observación morfológica in situ, resultó imposible y, sin material purificado, no era factible desarrollar ensayos serológicos. Fue en este caso cuando se utilizó por primera vez el ADNr 16S para la identificación, independiente de cultivo, de un agente patógeno17. El ADNr 16S del agente causal de la angiomatosis bacilar se amplificó a partir de tejido infectado, utilizando oligonucleótidos deducidos en base a regiones conservadas, comunes a todas las bacterias. El análisis de la secuencia amplificada reveló una nueva bacteria, estrechamente relacionada con Rochalimaea (después Bartonella) quintana. La identificación permitió poco después idear condiciones para su cultivo en el laboratorio18 y esto, a su vez, permitió el desarrollo de ensayos serológicos. La estrategia que condujo a la identificación de B. quintana fue posteriormente adoptada como método general para el descubrimiento de nuevos agentes patógenos19.
Se ha utilizado la misma metodología para la identificación directa de agentes infecciosos, a partir de muestras clínicas que deberían encontrarse estériles, sin necesidad de cultivo previo. Así, se han identificado bacterias patógenas presentes en el líquido cefalorraquídeo20,21, líquido sinovial22, válvulas cardíacas23 y sangre24-26. Aunque la identificación directa a partir de muestras clínicas es una técnica sumamente eficaz, carece de algunas de las ventajas que ofrece el cultivo11. Por ejemplo, en ausencia de éste, no es posible la caracterización posterior del microorganismo infeccioso, incluida la determinación de su susceptibilidad a agentes antimicrobianos o la diferenciación intraespecie para estudios epidemiológicos. Por ello, la identificación directa a partir de muestras clínicas se utiliza principalmente cuando fracasa el cultivo.
Actualmente, una de las aplicaciones más importantes del ADNr 16S es la identificación de micobacterias no tuberculosas, cuya incidencia y relevancia clínica ha aumentado de manera considerable en la última década, asociada a la pandemia del sida. El método tradicional de identificación de estas micobacterias se basa en características fenotípicas (pruebas bioquímicas, producción de pigmento, fisiología, y morfología de las colonias), cuya determinación requiere un tiempo considerable, como consecuencia del crecimiento lento de estas bacterias en medios de cultivo convencionales. Además, la interpretación de los resultados exige experiencia, y está limitada por la baja especificidad y la subjetividad. Por ello, la secuenciación del ADNr 16S se está imponiendo como método rápido y preciso para la identificación tanto de micobacterias previamente reconocidas como de nuevas especies13,15,27-29. La identificación molecular puede completarse 1 o 2 días después del aislado en cultivo puro, y puede incluso realizarla personal sin experiencia en biología molecular.
El sistema MicroSeq500, utilizado en la mayor parte de los trabajos ya citados, se está aplicando también a la identificación de bacterias corineformes, grupo para el cual los métodos fenotípicos disponibles en los laboratorios de microbiología clínica son sustancialmente deficientes. Tang et al14 analizaron un total de 52 aislados, y encontraron que a nivel de género los resultados de la identificación por secuenciación eran concordantes con los obtenidos utilizando criterios fenotípicos. A nivel de especie, la identificación por secuenciación fue más fiable en las dos especies de Corynebacterium con mayor relevancia clínica: C. diphteriae y C. jeikeium. El tiempo requerido fue inferior a 24 h.
Por último, y como ejemplo de aplicación a la identificación de bacterias con perfiles bioquímicos ambiguos, cabe mencionar el trabajo de Woo et al30. Estos autores utilizaron el sistema MicroSeq500 para la identificación de 37 aislados bacterianos, clínicamente relevantes, que incluían bacterias aerobias grampositivas, bacterias anaerobias gramnegativas y especies del género Mycobacterium, previamente identificadas por métodos fenotípicos. El 81,1% de los aislados pudieron ser correctamente identificados, mientras que en el 13,5% de los casos la identificación de género fue incorrecta y en el 5,4% (2 de 37), de especie. En 5 casos, la identificación errónea se debió a la ausencia de las secuencias ADNr 16S de las bacterias correctas en la base de datos del sistema MicroSeq500. Estos resultados apoyan la conveniencia de utilizar más de una base de datos, sobre todo cuando se trata de bacterias que raramente se encuentran en la práctica clínica.
Conclusión
La secuenciación del ADNr 16S constituye un método rápido y eficaz de identificación bacteriana, del cual puede beneficiarse la microbiología clínica, al igual que otras ramas de la microbiología. A medida que los recursos técnicos aumentan, el precio se hace más competitivo, por lo que probablemente su utilización para la identificación sistemática de bacterias patógenas será sólo cuestión de tiempo.
Agradecimientos
Queremos agradecer a los Dres. Fernando Vázquez del Hospital Monte Naranco de Oviedo, M. Cruz Martín, del Instituto de Productos Lácteos (Asturias) y José Luis Martínez Fernández, del Servicio de Secuenciación Automática de la Universidad de Oviedo, por la lectura crítica de este artículo, y por sus comentarios.
NOTA
Los artículos publicados en la sección "Formación Médica Continuada" forman parte de grupos temáticos específicos (antibiograma, antimicrobianos, etc.). Una vez finalizada la publicación de cada tema, se irán presentando al Sistema Español de Acreditación de la Formación Médica Continuada (SEAFORMEC) para la obtención de créditos.
Una vez concedida la acreditación, ésta se anunciará oportunamente en la revista y se abrirá un período de inscripción gratuito durante 3 meses para los socios de la SEIMC y suscriptores de la Revista, al cabo del cual se iniciará la evaluación, durante 1 mes, que se realizará a través de la web de Ediciones Doyma.
ANEXO 1. Identificación bacteriana mediante secuenciación del ARNr 16S: fundamento y aplicaciones en microbiología clínica
1. La utilización de ácidos nucleicos y proteínas como cronómetros moleculares, fue inicialmente propuesta por:
a)Woese.
b)Zuckerkandl y Pauling.
c)Pasteur.
d)Darwin y Haeckel.
e)Whittaker.
2. Las macromoléculas más ampliamente utilizadas en estudios filogenéticos y taxonómicos son:
a)Los ARNr 5S y 16S.
b)Los ARNr 18S y 23S.
c)Los ARNr 16S y 23S.
d)Los ARNr 16S y 18S.
e)Las proteínas ribosómicas.
3. Los organismos celulares se agrupan actualmente en los siguientes dominios:
a) Bacteria, Plantae, Animalia.
b) Virus, Prokarya, Eucarya.
c) Bacteria, Archaea, Eukarya.
d) Prokaryotae, Protista, Fungi, Plantae, Animalia.
e) Prokarya y Eukarya.
4. La utilización del ARNr 16S como cronómetro molecular tiene en cuenta que:
a)Se encuentra presente en todas las bacterias.
b)Su estructura y función han permanecido constantes a lo largo del tiempo.
c)Contiene regiones constantes y variables.
d)Existen bases de datos que incluyen un número muy elevado de secuencias.
e)Todas las anteriores.
5.El operón ribosómico (rrn) de las bacterias:
a)Puede encontrarse en más de una copia por genoma.
b)Contiene genes para los ARNr 16S, 18S y 23S.
c)Contiene genes para ARNs y proteínas ribosómicas.
d)Sólo contiene genes para ARNs ribosómicos.
e)Cada gen del operón se transcribe independientemente.
6. En taxonomía bacteriana la especie se define como:
a)Conjunto de cepas que comparten una similitud del 70% o más en experimentos de reasociación ADN-ADN.
b)Conjunto de cepas cuyos ADNs 16S comparten una identidad del 93% o más.
c)Conjunto de cepas pertenecientes al mismo serotipo.
d)Conjunto de cepas que pueden intercambiar información genética mediante recombinación homóloga.
e)Todas las anteriores.
7. En la identificación por secuenciación de ADNr 16S, la utilización de más de una base de datos puede ser decisiva cuando:
a)El amplicón secuenciado procede directamente de una muestra clínica.
b)Se utilizan bases de datos, de libre acceso o comerciales, potencialmente incompletas.
c)Se desea una identificación rápida.
d)Se pretenden construir árboles filogenéticos.
e)Se comparan secuencias de especies conocidas.
8. La primera vez que la secuenciación del ADNr 16S condujo al descubrimiento de un nuevo agente patógeno el método se aplicó a muestras clínicas de pacientes con angiomatosis bacilar. La bacteria identificada fue:
a) Tropheryma whipellii.
b) Francisella turalensis.
c) Haemophilus ducreyi.
d) Bartonella quintana.
e) Pseudomonas aeruginosa.
9. La identificación directa de bacterias en muestras clínicas mediante secuenciación del ADNr 16S respecto a la identificación a partir de cultivo tiene como limitación:
a)Es más costosa.
b)Requiere especialistas en genética molecular.
c)No se aísla la bacteria, por lo que no es posible la caracterización posterior.
d)No sirve para bacterias emergentes como patógenos humanos.
e)No permite identificar bacterias responsables de bacteremias.
10. En la actualidad, una de las aplicaciones más importantes de la secuenciación del ADNr 16S es la identificación de:
a)Especies con baja relevancia clínica.
b)Especies resistentes a antimicrobianos.
c) Pseudomonas aeruginosa.
d) Neisseria meningitidis.
e) Micobacterias no-tuberculosas.

