




Varón de 26 años con antecedente de esplenectomía tras traumatismo toracoabdominal 10 años atrás, que consultó a urgencias por fiebre de 4h de evolución, asociando malestar general, cefalea, mareo y vómito. Durante su estancia en urgencias presentó descompensación hemodinámica, con posterior shock séptico y fallo multiorgánico, por lo cual ingresó en la unidad de cuidados intensivos (UCI). En el estudio analítico, además de elevación de reactantes de fase aguda, acidosis metabólica y alteración de la función hepática y renal, presentaba signos compatibles con coagulopatía (prolongación de tiempos de coagulación, disminución de fibrinógeno y trombocitopenia), sin encontrarse evidencia clínica, analítica ni en pruebas de imagen de foco infeccioso.
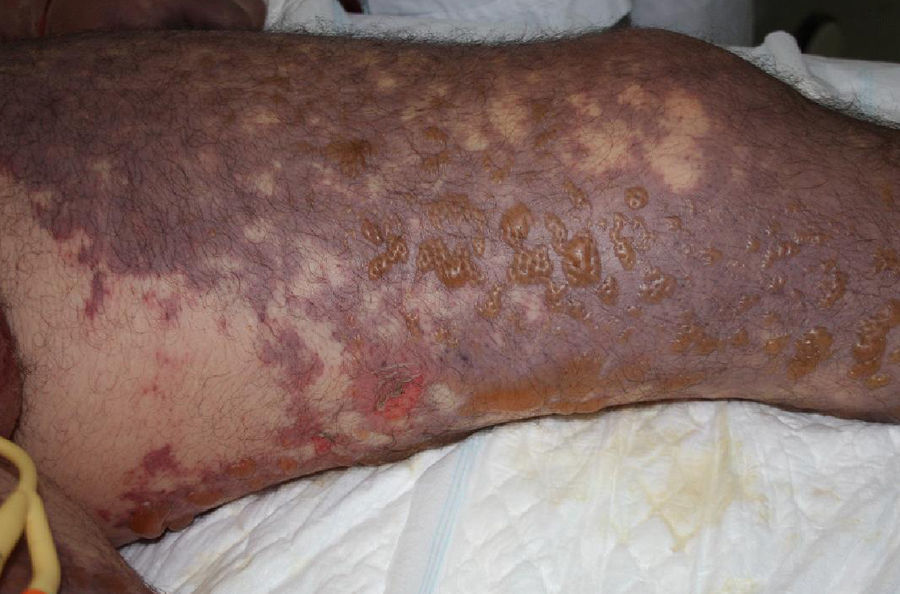
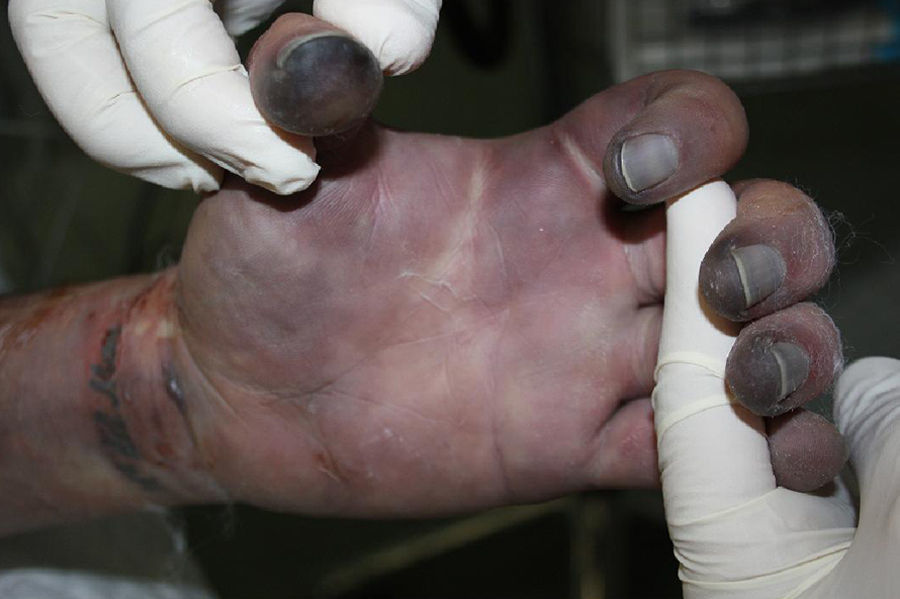
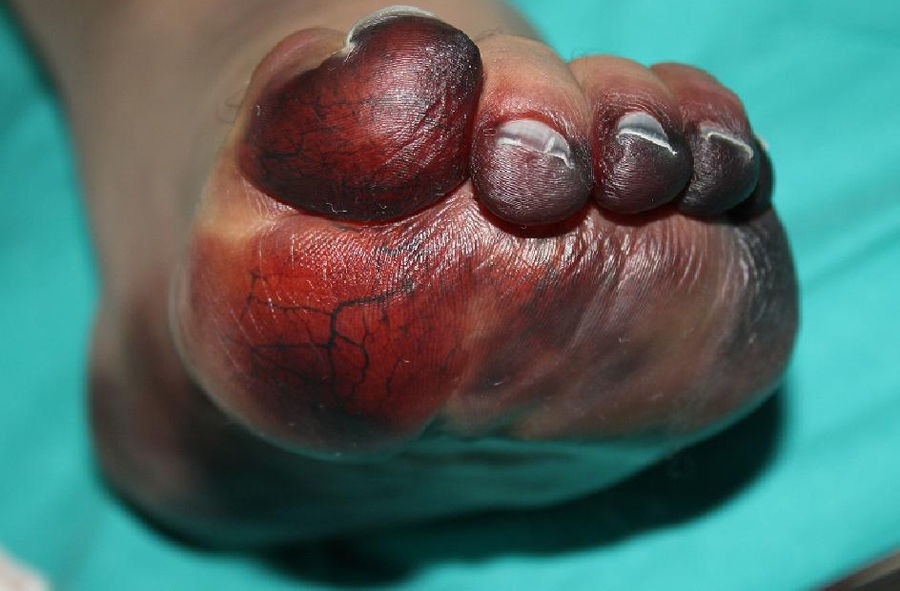
Al ingreso se observaba cianosis en zonas acras, con progresivo desarrollo en las siguientes horas de máculas purpúricas reticuladas de distribución simétrica en las 4 extremidades, mejillas y abdomen. Un día después presentaba áreas extensas de despegamiento epidérmico y formación de ampollas sobre las lesiones purpúricas previas (fig. 1), asociando frialdad distal y disminución de pulsos periféricos. Dos días más tarde desarrolló áreas grisáceo-negras de aspecto necrótico en manos y pies, con dedos afilados de consistencia pétrea (figs. 2 y 3).
Diagnóstico y evolución


Desde el momento del ingreso se inició terapia antibiótica empírica de amplio espectro con piperacilina/tazobactam; requiriendo además el suministro de fármacos vasoactivos, transfusión de hemoderivados y sedoanalgesia. Ante el aislamiento de Streptococcus pneumoniae en hemocultivos se modificó el tratamiento antibiótico pautándose meropenem, logrando una progresiva estabilización hemodinámica, con persistencia de áreas de necrosis en zonas acras, lo que condujo finalmente a la amputación quirúrgica de las 4 extremidades a diferentes niveles.
ComentarioLa purpura fulminans (PF) es una patología grave de inicio agudo, rápida progresión y alta morbimortalidad, caracterizada por la aparición de extensas áreas de necrosis cutánea en presencia de coagulación intravascular diseminada (CID)1.
La PF se presenta en 3 contextos clínicos: a)déficit congénito de proteína C o S (PF neonatal); b) déficit transitorio autoinmune de proteína C o S (PF idiopática), y c)CID en el contexto de una infección aguda grave (PF infecciosa)2.
La PF infecciosa se asocia principalmente a sepsis por Neisseria meningitidis; sin embargo, S. pneumoniae se comporta como un agente etiológico muy común en población esplenectomizada3. La PF infecciosa se caracteriza por la presencia de máculas violáceas que progresan hacia la formación de vesículas y ampollas, con rápida aparición de áreas de necrosis cutánea. Se presenta en el contexto de una respuesta inflamatoria aguda en casos de sepsis severa, en la cual hay una activación sistémica de la cascada de la coagulación. Esto promueve la formación de microtrombos en vasos dérmicos con isquemia endotelial, lo cual, asociado al posterior consumo de factores de coagulación, resulta en necrosis cutánea hemorrágica4.
El tratamiento de esta patología se enfoca en el soporte hemodinámico y la cobertura antibiótica precoz dirigida hacia el posible agente etiológico causal, teniendo en cuenta las posibles resistencias terapéuticas5. En el caso de pacientes asplénicos se debe sospechar la infección por bacterias encapsuladas6. En infecciones confirmadas por S. pneumoniae el tratamiento de elección es un fármaco betalactámico. El diagnóstico temprano y el rápido inicio de tratamiento permiten reducir la mortalidad y disminuir la morbilidad asociada a la pérdida de tejidos por necrosis7.

