





Rilpivirina es un potente inhibidor no nucleósido de la transcriptasa inversa que ha mostrado gran eficacia en el tratamiento de la infección por el virus de la inmunodeficiencia humana 1 (VIH-1) en pacientes naïve. Rilpivirina es un fármaco activo, tanto frente a cepas salvajes de VIH-1 como frente a una extensa variedad de cepas virales resistentes a los inhibidores no nucleósidos de la transcriptasa inversa de primera generación. Posee un perfil farmacocinético muy favorable, aunque al ser su absorción dependiente del pH gástrico debe ser administrada con comida para asegurar su correcta absorción. Su metabolismo está mediado a través del citocromo P450 (CYP) 3A, por lo que deben considerarse las potenciales interacciones cuando se administre conjuntamente con inductores o inhibidores de esta vía enzimática. Aunque a dosis más altas puede comportarse como inductor enzimático, no es esperable que rilpivirina a la dosis de 25mg al día pueda alterar las concentraciones de otros fármacos metabolizados por esta vía. Su prolongada vida media permite su administración por vía oral 1 vez al día.
Rilpivirine is a potent nonnucleoside reverse transcriptase inhibitor (NNRTI) with high efficacy in the treatment of HIV infection in treatment-naïve patients. This drug is active against both wild-type HIV-1 and a wide variety of first-generation NNRTI. Rilpivirine has a highly favorable pharmacokinetics profile, but, because its absorption depends on gastric pH, it should be administered with food to ensure correct absorption. Rilpivirine is metabolized by cytochrome P450 (CYP) 3A and consequently potential interactions should be considered when it is administered with P450 (CYP) 3A inducers or inhibitors. Although higher doses can behave as enzyme inducers, at a dose of 25mg/day, rilpivirine is unlikely to alter the concentrations of other drugs metabolized through this pathway. Because of its prolonged half-life, rilpivirine can be administered orally once daily.
Rilpivirina (RPV) es un nuevo fármaco antirretroviral perteneciente a la familia de los inhibidores no nucleósidos de la transcriptasa inversa (INNTI), con una eficacia demostrada en pacientes naïve y una excelente tolerabilidad. Comparte con efavirenz (EFV) alguna de sus beneficiosas características farmacocinéticas, como una vida media muy prolongada, y también las limitaciones relacionadas con su metabolización a través del citocromo P450. En el presente artículo expondremos el mecanismo de acción y las principales características farmacocinéticas de RPV.
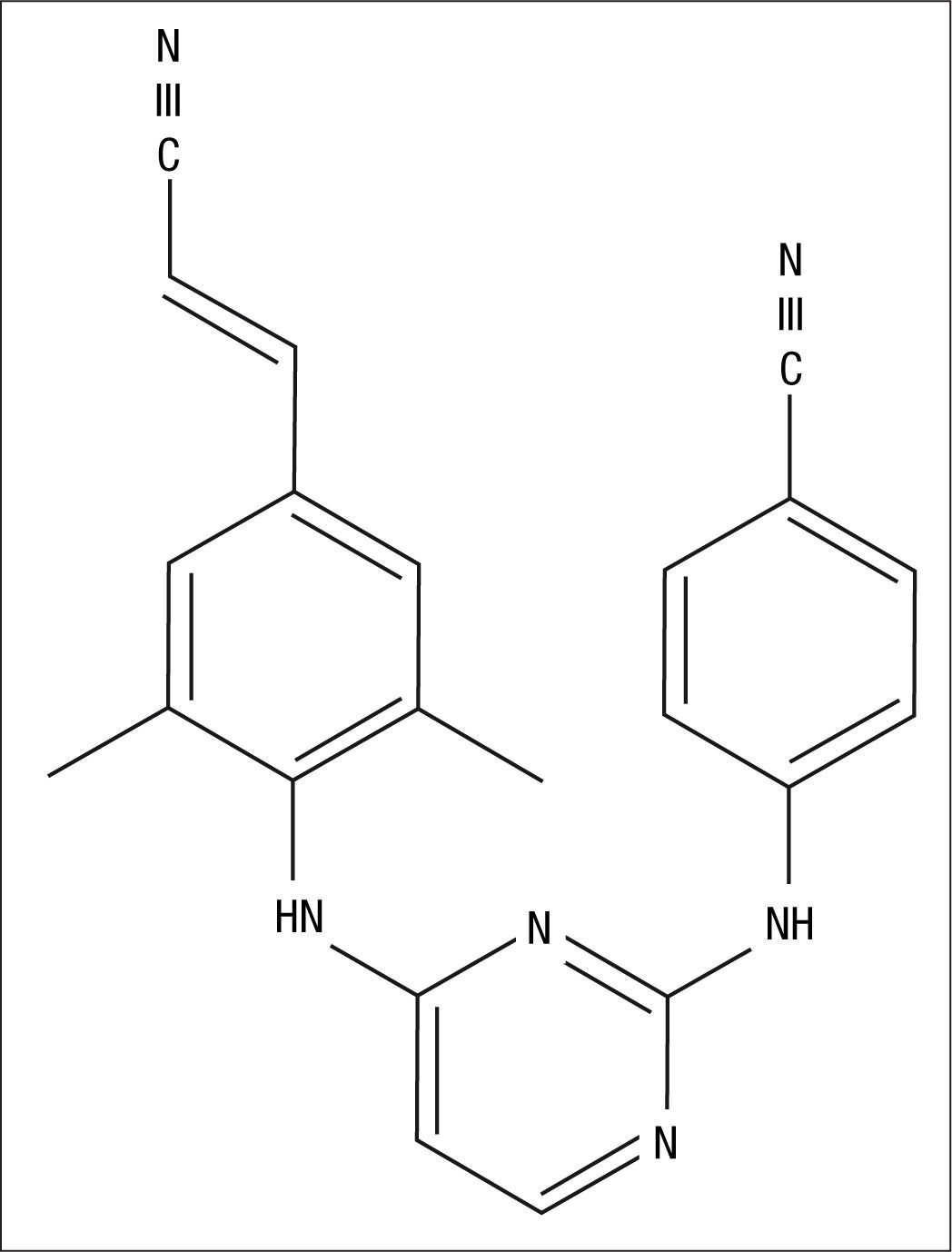
Mecanismo de acciónRPV es un INNTI diarilpirimidínico (fig. 1) que inhibe la transcriptasa inversa del virus de la inmunodeficiencia humana 1 (VIH-1) mediante un mecanismo no competitivo1–3. Al igual que el resto de INNTI se une a la transcriptasa inversa cerca del sitio activo de la enzima, en el interior de un pequeño “bolsillo” hidrofóbico. RPV induce cambios en la conformación de la enzima dependiendo de la estructura química específica de esta, su tamaño y de su forma de unión4. Estos cambios conformacionales inhiben la catálisis del ARN-viral y su transformación en ADN-viral que realiza la transcriptasa, consiguiendo disminuir la capacidad replicativa del VIH-1.

RPV no inhibe las ADN polimerasas humanas α, β o γ5 y, por lo tanto, no se asocia a la toxicidad mitocondrial asociada a los inhibidores de la transcriptasa inversa análogos de nucleósidos (ITIAN)6.
Los INNTI de segunda generación, etravirina (ETR) y RPV, poseen una estructura flexible mediante la cual consiguen adaptarse a los posibles cambios existentes en el sitio específico donde se unen a la enzima, incluso cuando hay cambios derivados por la existencia de mutaciones genéticas de resistencia que confieren resistencia a los INNTI de primera generación, EFV y nevirapina (NVP)7,8. La eficacia inhibitoria de RPV y ETR sobre la transcriptasa inversa mutante se debe a su elevada flexibilidad entre los anillos aromáticos, que les confiere la plasticidad necesaria para adoptar múltiples conformaciones9. EFV y NVP se unen preferentemente a los sitios Y181 e Y188 localizados en la región “palm” de la transcriptasa inversa. ETR y RPV se unen al mismo bolsillo, pero dependen más de otros puntos de unión, como por ejemplo la localización W229, que presenta una baja tasa de mutaciones10. Por esta razón, la mutación que con más frecuencia se asocia a resistencia a los INNTI de primera generación, la K103N, no compromete la eficacia de RPV11. El perfil de resistencia a RPV se expondrá en otro capítulo de esta monografía.
FarmacocinéticaLos datos farmacocinéticos de RPV disponibles (tabla 1) para la dosis recomendada de 25mg/día por vía oral (v.o.) provienen de 3 estudios realizados en pacientes con infección por VIH-1 naïve al tratamiento antirretroviral. El primero de ellos es un estudio que utilizó dosis múltiples escaladas (n=8)12, un ensayo clínico en fase IIb, aleatorizado con dosis de RPV diferentes (n=89)13 y un estudio farmacocinético poblacional que utilizó muestras de los estudios pivotales de RPV en fase III (n=679)14. El resto de datos se han obtenido de estudios en voluntarios sanos sin infección por VIH, con dosis de RPV marcada radiactivamente15.
Perfil farmacocinético y farmacodinámico de rilpivirina.
| Mecanismo de acción | |
| Clase farmacológica | INNTI de 2.a generación |
| Mecanismo de acción | Inhibición no competitiva de la transcriptasa inversa del VIH-1 |
| Actividad antiviral in vitro | Activa frente a: |
| – VIH-1 cepa wild-type | |
| – Subtipos A, B, C, D, F, G, H (grupo M) | |
| – VIH-1 con mutación K103N | |
| – VIH-1 con otras mutaciones asociadas a resistencia a EFV o NVP | |
| Menor actividad frente a VIH-1 grupo O | |
| No actividad frente a VIH-2 | |
| Dosis y administración | |
| Dosis | 25mg |
| Vía de administración | Oral |
| Frecuencia de administración | Una vez al día |
| Forma de administración | Con comida |
| Farmacocinética de RPV | |
| Absorción v.o. | – Dependiente del pH |
| – Absorción: 40% en ayunas | |
| – Absorción: 50% con bebida hiperproteica | |
| Distribución | Unión a proteínas: 99,7% |
| Biotransformación | Metabolismo oxidativo |
| Sustrato e inductor del CYP 3A4 | |
| Eliminación | Heces y orina |
| En orina < 1% de RPV activa | |
| Parámetros farmacocinéticos de RPV 25mg v.o. administrada con comida | |
| Concentración plasmática 24h — media— | 80,0ng/ml |
| AUC concentración plasmática -media- (0–24h) | 2.397ng-h/ml |
| Tiempo hasta la Cmax plasmática | 4–5h |
| Aclaramiento oral | 11,8l/h |
| Vida media de eliminación | ≈45h |
AUC: área bajo la curva; Cmax: concentración máxima; EFV: efavirenz; INNTI: inhibidor no nucleósido de la transcriptasa inversa; NVP: nevirapina; RPV: rilpivirina; VIH: virus de la inmunodeficiencia humana; v.o.: vía oral.
Tras la administración oral de RPV, la concentración plasmática máxima de RPV se alcanza en un plazo de 4–5h. Tras 7 días de RPV a una dosis diaria de 25mg, la concentración máxima alcanzada (Cmax) fue de 263ng/ ml, la media del área bajo la curva (AUC) en plasma de 0 a 24h (AUC24) fue de 3.659ng/h/ml y la mediana de tiempo hasta alcanzar la Cmax fue de 4h. RPV es detectable en plasma en la mayoría de los pacientes hasta 168h después de la última dosis.
La absorción de RPV es dependiente del pH, incrementándose su biodisponibilidad en un ambiente ácido. Cuando se coadministra con antiácidos o inhibidores de la secreción ácida gástrica se observa una reducción significativa en la absorción de RPV. Por ello, RPV no debe coadministrarse con los inhibidores de la bomba de protones. Si se utilizan antiácidos o antagonistas de los receptores H2, la administración de ambos fármacos debe separarse suficientemente en el tiempo.
RPV siempre se deberá administrar con comida para asegurar su correcta absorción. La exposición a RPV es aproximadamente un 40% menor cuando se administra en ayunas, en comparación con una comida calórica normal (533kcal), o con una comida hipercalórica y rica en grasas (928kcal). Estos datos indican que no es necesaria una dieta hipercalórica y grasa para asegurar la absorción del fármaco. Sin embargo, cuando RPV se administra solo con una bebida nutricional rica en proteínas, la exposición a RPV disminuye en un 50% respecto a cuando se toma conjuntamente con una comida. Por todo ello se recomienda que RPV se tome con una comida para lograr una absorción óptima. La administración de RPV en ayunas o con una bebida nutricional hiperproteica reduce sus concentraciones plasmáticas, lo que podría disminuir su eficacia.
DistribuciónRPV se une en un 99,7% a las proteínas plasmáticas, principalmente a la albúmina. No se conoce la distribución de RPV en otros compartimientos diferentes al plasma como, por ejemplo, líquido cefalorraquídeo, secreciones genitales, etc.
Relación farmacocinética-farmacodinámicaEn el estudio de búsqueda de dosis se observó un modesto impacto de las concentraciones plasmáticas de RPV en la respuesta clínica (CVP [carga viral plasmática]-VIH < 50 copias/ml). De los pacientes que recibieron RPV a dosis de 25–150mg al día, respondió el 82,8% de los pacientes en el cuartil más bajo de exposición al fármaco (AUC24 plasmática < 3.023ng/h/ml), frente al 91,1–93,2% de los pacientes en los cuartiles más elevados de AUC24. A pesar de ello, no se observaron diferencias en la eficacia viral entre las diferentes dosis administradas16.
BiotransformaciónLos estudios in vitro indican que RPV experimenta un metabolismo oxidativo mediado por el citocromo P450 (CYP) 3A. La RPV es sustrato e inductor del citocromo CYP3A4 y solo el 0,03% del fármaco activo es eliminado por la orina. Los fármacos que inducen o inhiben el CYP3A pueden modificar el aclaramiento de RPV.
EliminaciónLa semivida de eliminación terminal de RPV es de 45h, aproximadamente. Tras la administración v.o. de dosis únicas de 14C-rilpivirina, un promedio del 85 y del 6,1% de radioactividad se recupera en heces y orina, respectivamente. En las heces se detecta un promedio del 25% de la dosis administrada de RPV. En la orina se detectan únicamente cantidades mínimas de RPV intacta (< 1% de la dosis). En los estudios farmacocinéticos poblacionales, el aclaramiento oral aparente es de 11,8 l/h, con una variabilidad individual del 39%.
Farmacocinética de la coformulación de rilpivirina con tenofovir y emtricitabinaSe ha desarrollado una tableta única que combina a dosis fijas te-nofovir-disproxil-fumarato (300mg), emtricitabina (200mg) y RPV (25mg). Los datos farmacocinéticos de esta combinación se han obtenido de un estudio abierto, aleatorizado en voluntarios sanos (n=34) que comparó la farmacocinética de la coformulación con los mismos fármacos administrados en diferentes tabletas. En el estudio realizado se demostró la bioequivalencia de ambos tratamientos en las medias plasmáticas de la Cmax y las AUC obtenidas para cada uno de los fármacos cuando se daban de forma separada o coformulados. Se observó bioequivalencia para cada uno de los parámetros farmacocinéticos estudiados, con un intervalo de confianza del 90% de 80–12517.
Farmacocinética en poblaciones especialesPoblación pediátricaLa farmacocinética de RPV en pacientes pediátricos no está suficientemente estudiada, por lo que no hay datos suficientes en la actualidad para realizar recomendaciones en esta población.
Pacientes de edad avanzadaEl análisis farmacocinético poblacional realizado en pacientes con infección por VIH demostró que la farmacocinética de RPV no varía en los diferentes intervalos de edad analizados (18 a 78 años), con solo 2 pacientes de 65 años o más. No es necesario ajustar la dosis de RPV en esta población, aunque en ausencia de más datos se debe usar con precaución.
SexoAunque se observó un menor aclaramiento del fármaco en mujeres, dicha diferencia no se considera clínicamente relevante18.
RazaLos pacientes asiáticos tendrían un menor aclaramiento de RPV, pero esto no parece traducirse en una mayor toxicidad.
Insuficiencia hepáticaRPV se metaboliza y elimina fundamentalmente por vía hepática. En un ensayo en el que se compararon 8 pacientes con insuficiencia hepática leve (clase A de Child-Pugh) con 8 controles pareados y 8 pacientes con insuficiencia hepática moderada (clase B de Child-Pugh) con 8 controles pareados, la exposición a dosis múltiples de RPV fue un 47% mayor en pacientes con insuficiencia hepática leve y un 5% mayor en pacientes con insuficiencia hepática moderada. No se recomienda ajuste de dosis en pacientes con insuficiencia hepática moderada, aunque se recomienda la vigilancia estrecha de toxicidad por RPV. No hay datos en pacientes con insuficiencia hepática grave (clase C de Child-Pugh), por lo que no se recomienda su utilización en este escenario.
El análisis de farmacocinética poblacional en pacientes coinfectados por virus de la hepatitis B o C demostró que la coinfección por virus hepatotrópicos no tiene efectos clínicamente relevantes sobre la exposición a RPV.
Insuficiencia renalNo se ha estudiado la farmacocinética de RPV en pacientes con insuficiencia renal. La eliminación renal de RPV es insignificante, por ello no es necesario realizar ajuste de dosis en pacientes con insuficiencia renal leve o moderada. En pacientes con insuficiencia renal grave o nefropatía terminal se debe utilizar RPV con precaución, ya que la concentración plasmática puede aumentar debido a la alteración de la absorción, distribución o metabolismo del fármaco secundario a la insuficiencia renal. La combinación de RPV con un inhibidor potente del CYP3A únicamente se debe utilizar en pacientes con insuficiencia renal grave o nefropatía terminal si el beneficio es mayor que el riesgo.
RPV presenta una gran afinidad por las proteínas plasmáticas, por lo que no es probable que la hemodiálisis o la diálisis peritoneal incrementen su eliminación de manera significativa.
La coformulación tenofovir-emtricitabina-RPV no debe utilizarse en pacientes con insuficiencia renal moderada, grave o enfermedad renal terminal, y en todos los que requieren diálisis. Al contrario que RPV, tenofovir y emtricitabina se eliminan principalmente por vía renal mediante secreción tubular y filtrado glomerular.
Administración de rilpivirina por vía parenteralSe ha estudiado de forma experimental la farmacocinética de RPV en formulación como suspensión de nanopartículas que permitirían la administración de RPV por vía intramuscular (i.m.) o subcutánea (s.c.), consiguiendo concentraciones plasmáticas de RPV adecuadas y estables durante largos períodos, lo que permitiría su administración en intervalos más prolongados, de hasta 3 meses19. La administración i.m. o s.c. de nanopartículas de RPV en animales de experimentación mostró una biodisponibilidad cercana al 100% y una buena difusión al tejido linfático20. También se ha estudiado en voluntarios sanos, pero no en pacientes con infección por el VIH21. Se ha estimado que la administración de 600nm RPV por vía parenteral 1 vez al mes conseguiría concentraciones similares a la dosis de 25mg al día por v.o. Entre las potenciales utilidades de esta posología estarían la profilaxis pre-exposición o la prevención de la transmisión maternofetal del VIH.
Conflicto de interesesEl Dr. Joaquín Portilla ha recibido pagos por conferencias por parte de Abbott, BMS, Boehringer-Ingelheim, Gilead, Janssen, MSD, Roche y ViiV, y pagos por asesorías de Abbott, Gilead, Janssen y MSD.
El Dr. Vicente Estrada ha recibido financiación económica para proyectos de investigación de Janssen y Abbott, y pagos por asesoría por parte de BMS, Abbott, Gilead, Janssen y MSD.

