




Los datos actualmente disponibles de seguridad y tolerabilidad de rilpivirina proceden de la ficha técnica del producto, del ensayo clínico en fase IIb de búsqueda de dosis TMC278-C204, de los ensayos clínicos en fase III ECHO y THRIVE, y de los datos preliminares de los ensayos STaR y SPIRIT, con un total de 1.728 pacientes. Su comparador habitual ha sido efavirenz, y en todos los estudios se ha observado una menor incidencia y severidad de efectos adversos neuropsiquiátricos, un mejor perfil lipídico y un menor número de casos con elevación subclínica de transaminasas en los pacientes tratados con rilpivirina. Sin embargo, el número de pacientes coinfectados por virus B o C es relativamente pequeño para obtener conclusiones definitivas en este tipo de pacientes. Asimismo, la experiencia en pacientes con insuficiencia hepática leve o moderada es limitada, y no se dispone de datos de seguridad en pacientes con insuficiencia hepática avanzada.
Currently available data on the safety and tolerability of rilpivirine come from the product information document, a phase IIb, dose-finding clinical trial (TMC278-C204), the phase III ECHO and THRIVE clinical trials, and the preliminary data from the STaR and SPIRIT clinical trials, with a total of 1,728 patients. The comparator has usually been efavirenz. All studies have found a lower incidence and severity of neuropsychiatric adverse effects, a better lipid profile, and a lower number of patients with subclinical transaminase elevation in patients treated with rilpivirine. However, because of the relatively low number of patients coinfected with hepatitis B or C virus, definitive conclusions cannot be drawn. Similarly, experience in patients with mild or moderate liver failure is limited and there are no safety data in patients with advanced liver failure.
Entre los posibles efectos adversos (EA) de los inhibidores no nucleósidos de la transcriptasa inversa (INNTI) de primera generación, efavirenz (EFV) y nevirapina (NVP), se encuentran las reacciones de hipersensibilidad y exantemas; además EFV se caracteriza por sus efectos adversos neuropsiquiátricos y NVP puede dar lugar a hepatotoxicidad severa que requiere una estrecha vigilancia durante las primeras 18 semanas de tratamiento, sobre todo en pacientes con recuentos de CD4 elevados1,2. Rilpivirina (RPV) es una molécula con una eficacia similar a los INNTI de primera generación con una menor incidencia de EA3.
Actualmente, los datos disponibles de seguridad y tolerabilidad de RPV proceden de: a) la ficha técnica del producto3; b) de un ensayo clínico en fase II de búsqueda de dosis, en el que distintas dosis de RPV (25, 75 y 150mg/día, n=279) se compararon con EFV administrado junto con 2 análogos de nucleósidos (zidovudina [AZT] y lamivudina [3TC] o tenofovir [TDF] y emtricitabina [FTC])4; c) de los 686 pacientes que recibieron RPV en los ensayos clínicos en fase III ECHO (Early Capture HIV Cohort Study) y THRIVE (Transcriptase Inhibitors in Treatment-naive Aadults Infected with HIV-1)5–9 en pacientes sin tratamiento antirretroviral previo, en los que RPV (25mg/día) se comparó con EFV asociado a TDF/FTC en el primero de ellos, y a TDF/FTC o AZT/3TC o ABC/3TC en el segundo; d) de los datos preliminares presentados en congresos del estudio STaR (Single-Tablet Regimen), en el que se comparan RPV (n=392) y EFV, ambos junto con TDF y FTC coformulados en un solo comprimido, en pacientes sin tratamiento previo10, y e) del estudio SPIRIT (Switching boosted PI to Rilpivirine In Combination with Truvada as a Single Tablet Regimen), en el que en pacientes en tratamiento con 2 análogos de nucleós(t)idos y un inhibidor de la proteasa (IP) potenciado con ritonavir se evalúa una estrategia de simplificación a RPV/TDF/FTC (n=317) en un solo comprimido, frente a continuar con los análogos y el IP potenciado11.
En el ensayo en fase II, el seguimiento medio fue de 2 años (100 semanas) constatándose una menor frecuencia de EA grados 2 a 4 en las ramas de RPV, comparado con el brazo de EFV (el 20,4 frente al 37,1%; p<0,01), aunque la frecuencia de EA severos (RPV el 12,2% frente a EFV el 14,1%) o que motivaran la discontinuidad del tratamiento (RPV el 11,5% frente a EFV el 9%) fue similar en ambos brazos. La incidencia de EA neuropsiquiátricos estuvo presente en ambas ramas de tratamiento, pero con una incidencia menor en los pacientes tratados con RPV (el 48,7 frente al 72,1%; p<0,001), sobre todo en la incidencia de mareos y pesadillas, independientemente de la dosis de RPV. Asimismo, la incidencia de exantema relacionado con los fármacos en investigación fue menor en el brazo de RPV (el 0,4 frente al 7,9%; p<0,0001), mientras que las náuseas o molestias abdominales fueron igual de frecuentes en ambos brazos (el 4,3 frente al 8%; p>0,16)4.
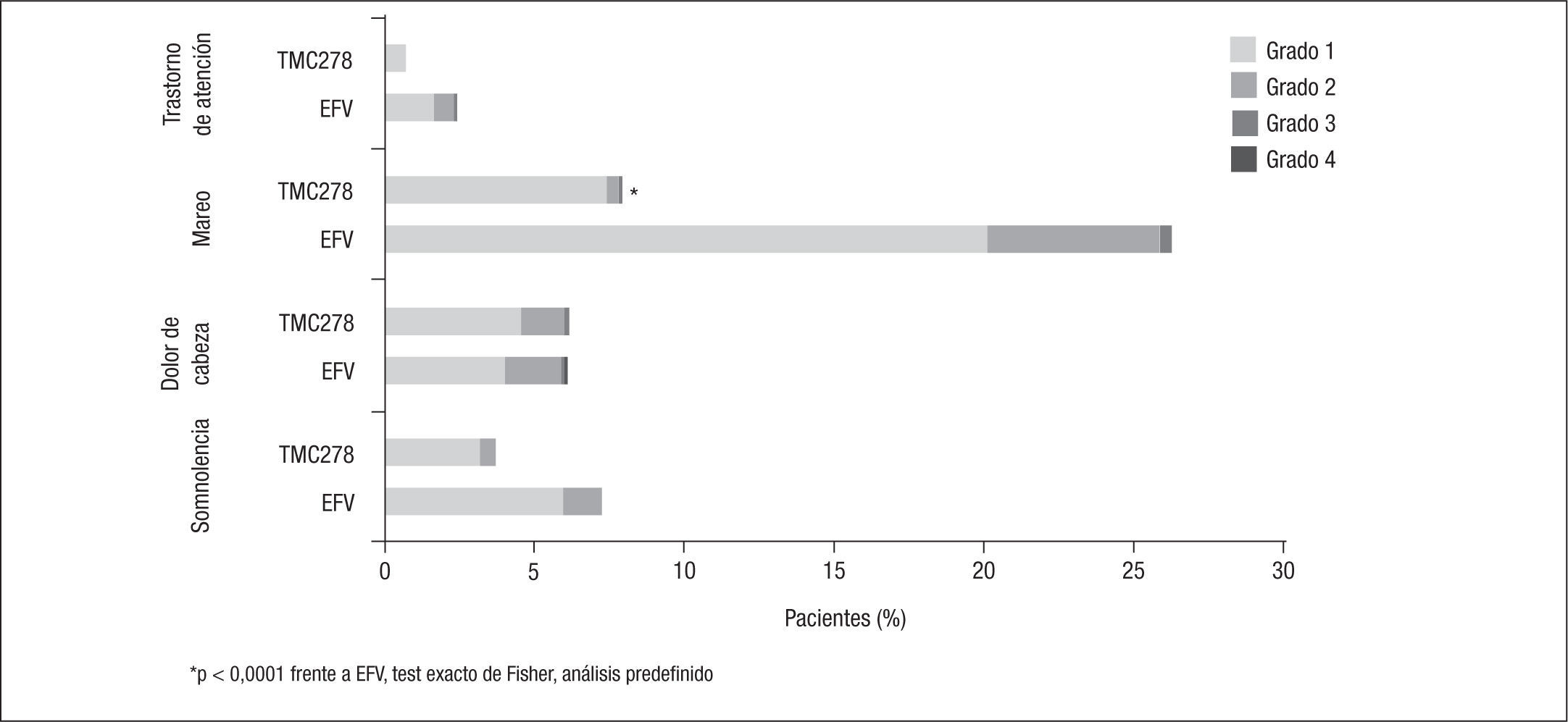
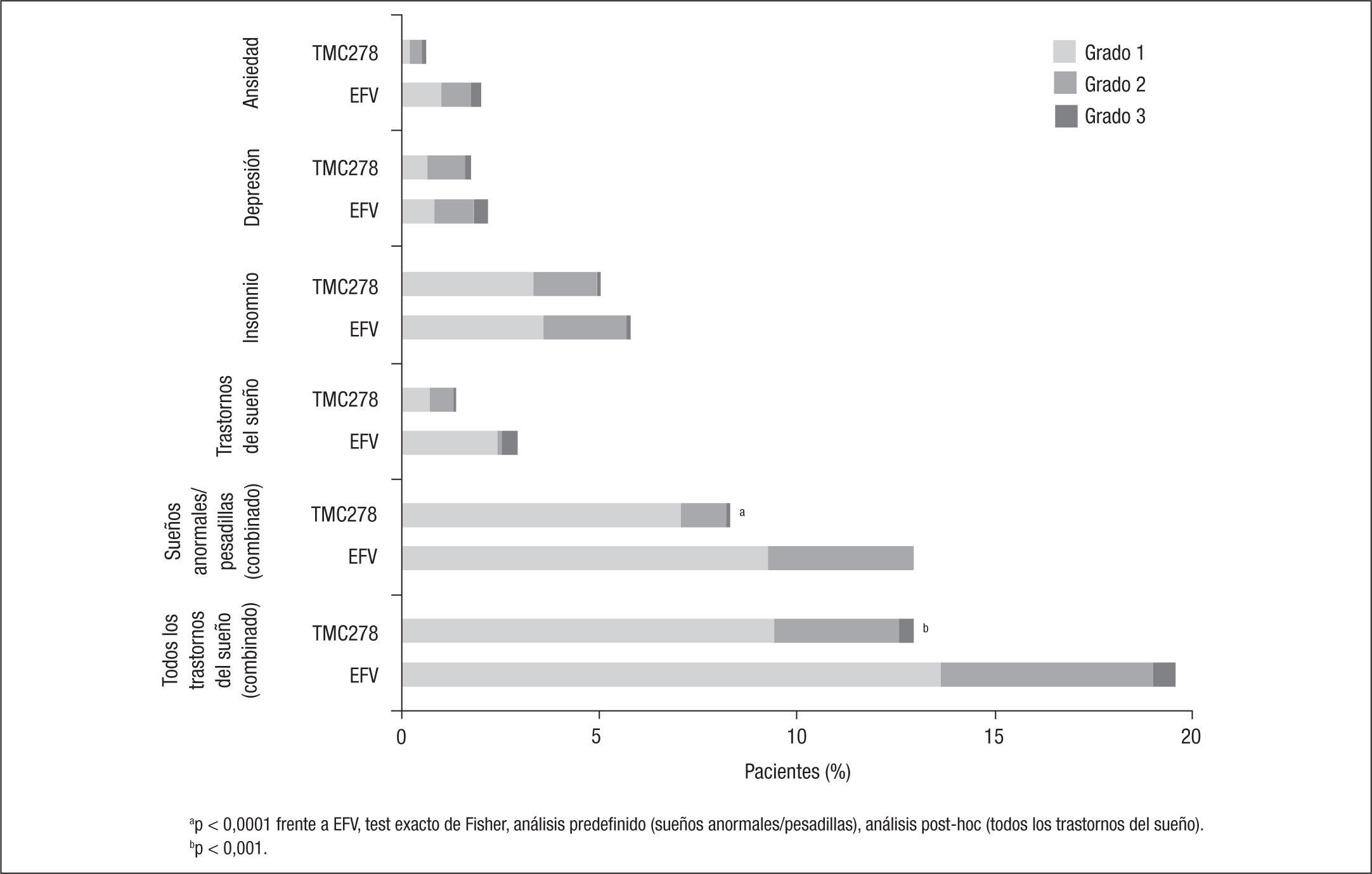
Dado que el diseño de los ensayos clínicos ECHO y THRIVE solo presentaba diferencias en las parejas de análogos utilizados en los lugares de reclutamiento de los pacientes, los resultados se suelen analizar conjuntamente5–9. En estos, la mayoría de los EA registrados fueron leves y ocurrieron en los primeros 3 meses, sobre todo en las primeras 4 semanas, con una frecuencia significativamente mayor de mareos (el 8,0 frente al 26,7%), pesadillas (el 8,3 frente al 13,2%) y exantema (el 4,2 frente al 15,1%) en el brazo de EFV respecto al de RPV. Más allá de las 48 semanas, el perfil de seguridad y tolerabilidad de ambos fármacos fue similar7. Aunque la incidencia de EA severos fue similar con RPV y EFV (el 9 y el 10%, respectivamente), con EFV fueron más frecuentes los EA que motivaron un cambio de tratamiento (el 9 frente al 4%; p<0,001) (figs. 1 y 2).

Incidencia de efectos adversos neurológicos de interés posiblemente relacionados con el tratamiento en ≥ 2% de los pacientes en cada grupo de tratamiento. Tomado de Mills et al9.

De los estudios STaR y SPIRIT, actualmente tan solo se dispone de información preliminar sobre la tasa de discontinuaciones de tratamiento por EA, siendo estas del 2 frente al 8% para los brazos de RPV y EFV, respectivamente, en el primero de ellos10 y del 1,5 frente al 0% para RPV frente al mantenimiento de los análogos junto con un IP/r en el segundo, con una tasa de EA grados 3–4 del 3,1% en el brazo de RPV11.
Otro de los EA preocupantes con cualquier régimen antirretroviral es la posibilidad de cambios en el tejido graso y la aparición de lipodistrofia. Aunque todavía no se conocen datos en este sentido derivados de los ensayos ECHO y THRIVE, in vitro se ha observado que tanto RPV como EFV inducen un efecto deletéreo sobre la diferenciación de los adipocitos asociada a una disminución de la expresión de distintos reguladores de la adipogénesis (PPAR-γ, C/EBP-α, SREBP-1), que actúan sobre los genes codificadores de lipoproteinlipasa, leptina y adiponectina, y aumentan la liberación de citocinas proinflamatorias (IL6, IL8, MCP-1 y PAI-1). Sin embargo, los efectos de EFV son mucho más acentuados que con RPV, precisándose dosis muy elevadas de este último para observar algunos de estos efectos12.
Aunque es muy improbable que a dosis terapéuticas de RPV (25mg/día) se observen interacciones farmacodinámicas con los medicamentos que prolongan el intervalo QTc en el electrocardiograma (ECG), en los ensayos de fase IIb, las dosis supraterapéuticas de RPV (75mg 1 vez al día y 300mg 1 vez al día) prolongaron el intervalo QTc en el ECG, por lo que se aconseja precaución cuando se administre conjuntamente con un fármaco asociado a riesgo de torsade de pointes3,13. Edurant® a la dosis recomendada de 25mg 1 vez al día no está asociado con un efecto clínicamente relevante sobre el intervalo QTc3.
Por otro lado, la toxicidad embrionaria y fetal de RPV solo se ha evaluado en ratas y conejos, con una exposición a RPV de 30 a 80 veces la observada en humanos con una dosis de 25mg al día, sin que se observara teratogenicidad y solo un paso limitado a placenta14. Sin embargo, su seguridad durante el embarazo no está establecida, ya que la gestación fue un criterio de exclusión en los ensayos clínicos con RPV. Por ello, RPV está clasificada como fármaco de categoría B por la Food and Drug Administration (no riesgo fetal en estudios animales, pero ausencia de estudios adecuados en mujeres embarazadas) para su uso durante el embarazo3.
Seguridad hepáticaHasta el momento, la mayoría de los datos proceden de los ensayos clínicos en fase III ECHO y THRIVE, con un seguimiento de 96 semanas, en los que la población coinfectada por los virus B o C de la hepatitis estaba poco representada, y también de los ensayos de fase II.
En el estudio TMC278-C204 en fase IIb, la proporción de pacientes coinfectados fue algo mayor que en los ensayos en fase III, con un 7% de coinfección por virus de la hepatitis C —VHC— (VHC-ARN+) y un 3% de coinfectados por VHB. Tras una mediana de seguimiento de 100 semanas se observó elevación de la AST grado 3–4 en el 3,3% de los pacientes y de la ALT en el 5,8% del grupo asignado a recibir 25mg de RPV, y globalmente fue del 6,6% entre los pacientes que recibieron RPV. Las alteraciones en la coagulación que probablemente serían de causa multifactorial se observaron con la dosis de 25mg en un 4,3% con prolongación de la TPTa, pero sin alteraciones del cociente internacional normalizado (INR). Entre los tratados con EFV hubo prolongación del TPTa en el 4,7% y del INR en el 2,3%4. Ya que se ha establecido la bioequivalencia entre el fármaco coformulado y sus componentes, los datos de seguridad de los diferentes estudios serían aplicables al fármaco coformulado15.
En los estudios ECHO y THRIVE, de los 1.335 pacientes incluidos en ambos ensayos, solo el 6 y el 9%, respectivamente, estaban coinfectados por VHB o VHC (HBsAg+ o ARN-VHC plasmático+). De ellos, 54 pacientes fueron aleatorizados al grupo de RPV y 3 se infectaron durante el estudio5,6.
La toxicidad analítica hepática, en las primeras 48 semanas de tratamiento, medida con la escala de los ACTG, fue inferior en los pacientes asignados a RPV, con una elevación de la ALT grado 3–4 del 1,5%, frente al 3,4% de los que recibieron EFV. En el caso de la AST fue del 2 y el 2,8%, respectivamente. Globalmente, la elevación de las enzimas hepáticas supuso un tercio de la toxicidad analítica severa.
Con un seguimiento mayor, entre las semanas 48 y 96 no se describieron nuevos tipos de toxicidad, y las alteraciones analíticas fueron moderadas y similares entre ambos tratamientos7.
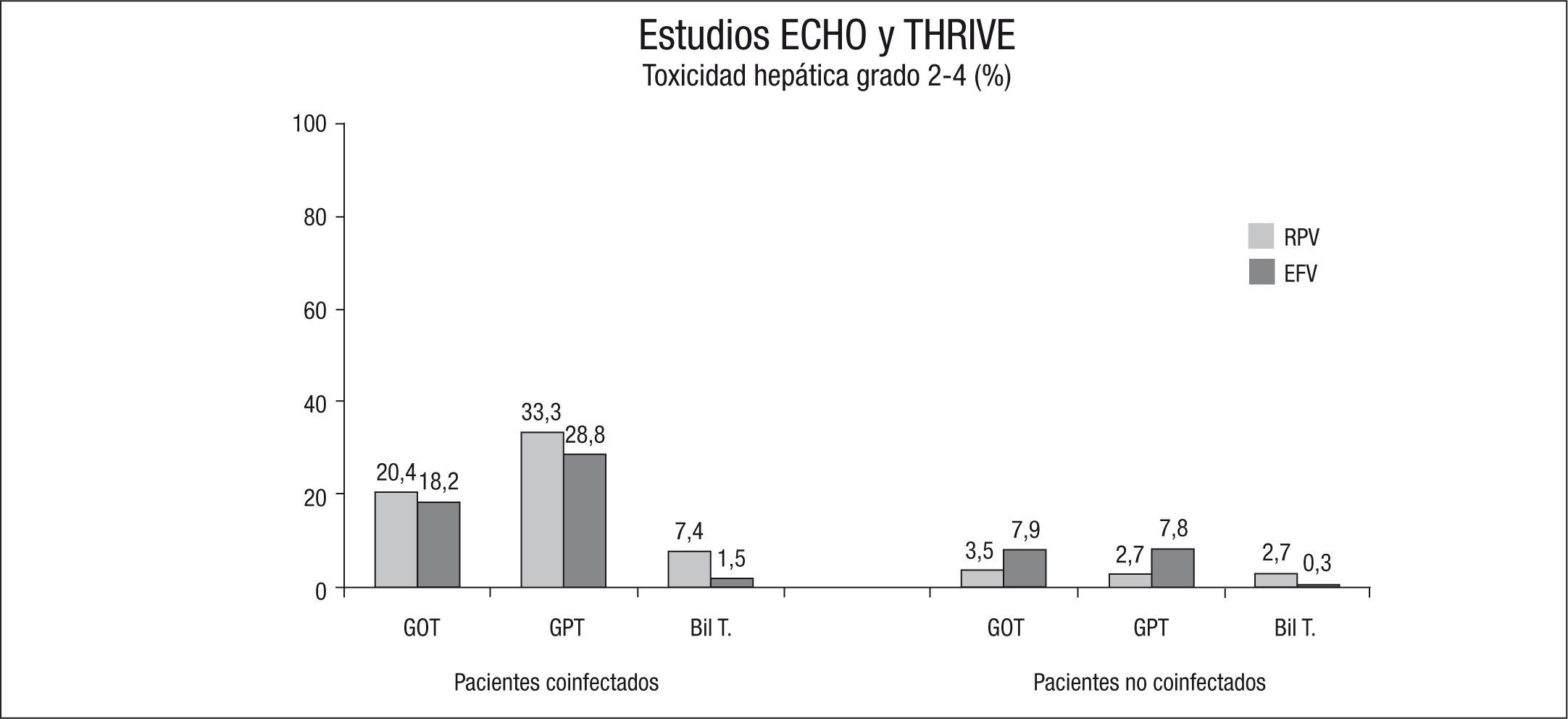
Las elevaciones de la ALT grados 2–4 se observaron entre los tratados con RPV en el 33,3% de los coinfectados frente al 2,7% en los no coinfectados y en el grupo de EFV en el 28,2 y 7,8%, respectivamente. Respecto a la AST, los porcentajes fueron del 20,4 y 3,5% en el grupo de RPV frente al 18,2 y 7,9% en el de EFV (fig. 3). Los 2 casos de efectos adversos hepáticos severos fueron solo analíticos, de grado 3 y ocurrieron en el grupo de EFV. En el grupo de RPV, 3 pacientes —2 de ellos coinfectados— interrumpieron el tratamiento por toxicidad hepática, frente a 9 en el grupo de EFV.

Hubo elevación de la fosfatasa alcalina grados 2–4 en el 7,4 frente al 2,5% en los pacientes con RPV coinfectados y no coinfectados y del 19,7 y 12,4% en el grupo de EFV. La incidencia de hiperbilirrubinemia grados 2–4 fue del 3,1% en el grupo de RPV frente al 0,4% en el de EFV (p=0,0003), fueron más frecuentes en los pacientes coinfectados, el 7,4 y 1,5% en los grupos RPV y EFV, y el 2,7 y 0,3% en los no coinfectados. No se observaron alteraciones de grado 4 y la elevación fue, sobre todo, a expensas de la fracción no conjugada. El mecanismo parece relacionado con el sistema de eliminación y no tendría que ver con toxicidad hepática ni con un aumento de la hemólisis.
Del estudio STaR no disponemos de datos definitivos, pero, como se ha comentado previamente, la toxicidad global grados 3–4 fue del 3,1%10.
En pacientes pretratados, los datos todavía preliminares provienen del estudio SPIRIT. La toxicidad hepática de grados 3–4 fue menor del 1% para cada alteración analítica evaluada11. Similar a otros estudios, la toxicidad hepática sería menos frecuente que en pacientes sin tratamiento previo.
En resumen, como sucede con otros antirretrovirales16,17, los pacientes coinfectados por VHB o VHC tratados con pautas que contienen RPV tienen un riesgo mayor de presentar toxicidad hepática que los pacientes monoinfectados por VIH en tratamiento antirretroviral. Sin embargo, la frecuencia de toxicidad severa es muy baja y la tolerabilidad en estos pacientes, muy buena18. En ninguno de los estudios comunicados hasta el momento se han descrito casos de hepatitis clínica.
Se ha descrito un caso de posible hepatitis alérgica por RPV en una paciente severamente inmunodeprimida, 4 días después de iniciar el tratamiento con una recuperación rápida tras la retirada del fármaco19.
No se dispone de datos de seguridad en pacientes con insuficiencia hepática avanzada. En pacientes con insuficiencia leve o moderada no se necesita ajustar la dosis y los valores del fármaco en el área bajo la curva (AUC) son un 47 y un 8% mayores. En todo caso, la experiencia en este escenario es muy limitada.
Perfil lipídicoAunque los INNTI como clase se han considerado tradicionalmente como fármacos con un perfil lipídico bueno, es preciso matizar esta afirmación. La comparación de los no análogos de nucleósidos NVP o EFV con los IP de primera generación, que se usaban sin potenciar con ritonavir, en general mostró un perfil lipídico más favorable para aquellos; en concreto, una menor elevación de triglicéridos, colesterol total y colesterol unido a lipoproteínas de baja densidad (cLDL), así como una mayor elevación del colesterol unido a lipoproteínas de alta densidad (cHDL)20. La comparación con los IP potenciados que se utilizan en la actualidad mostró mayores incrementos de triglicéridos, mientras que los parámetros de colesterol eran similares entre los fármacos de ambas clases o incluso superiores en NVP o EFV21–23. Posteriormente, etravirina mostró un menor impacto lipídico que EFV24, de forma que etravirina actualmente se considera un fármaco con un perfil lipídico neutro, al igual que los fármacos de otras familias como raltegravir25, elvitegravir26,27 o maraviroc28.
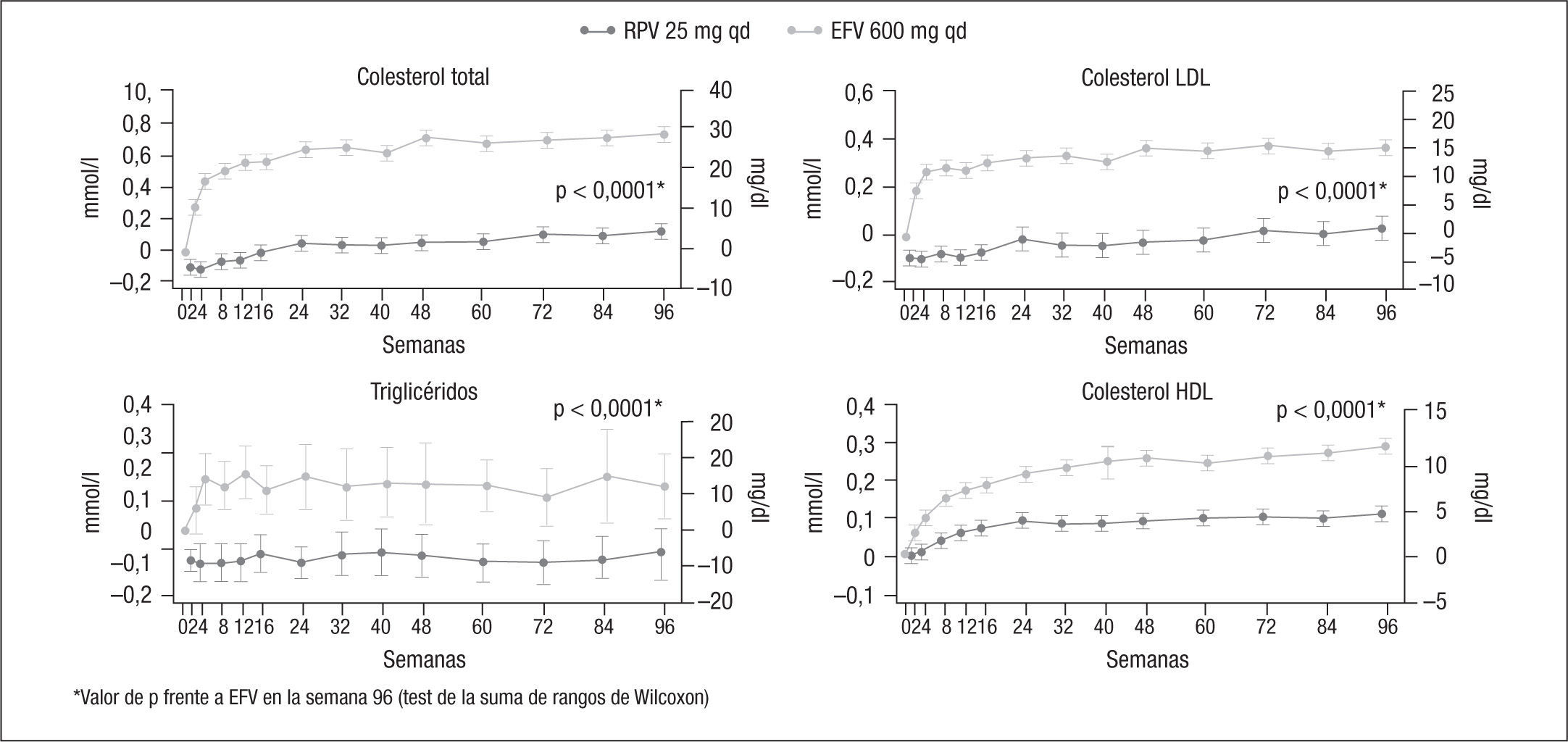
RPV tiene un comportamiento lipídico similar al de etravirina. En un estudio fase IIb, RPV a dosis ciegas de 25, 75 o 150mg 1 vez al día comparada con EFV 600mg 1 vez al día junto con AZT/3TC o TDF/FTC, el grupo de RPV tuvo un incremento de colesterol total de 9,0mg/dl (frente a 35,4mg/dl del grupo de EFV), de cLDL de 4,5mg/dl (frente a 18,2mg/dl del grupo de EFV), de cHDL de 6,2mg/dl (frente a 11,3mg/dl del grupo de EFV), y una reducción de triglicéridos de 9,9mg/dl (frente a un incremento de 29,2mg/dl del grupo de EFV) al cabo de 96 semanas29. Después de las 96 semanas, todos los pacientes de las ramas de RPV pasaron a tomar 75mg al día de forma abierta desde la semana 96 a la semana 144 y, posteriormente, a 25mg al día hasta la semana 192, y los valores lipídicos previamente comunicados para los pacientes tratados con RPV se mantuvieron de la misma forma en relación con los pacientes tratados con EFV11.
En el ensayo clínico THRIVE6,7,9, tras 96 semanas de seguimiento, el cambio medio de colesterol total fue de 3mg/dl en la rama de RPV y de 30,5mg/dl en la rama de EFV (p<0,0001), el cambio medio de cLDL fue de −0,7mg/dl en la rama de RPV y de 17mg/dl en la rama de EFV (p<0,0001), el cambio medio de cHDL fue de 4,2mg/dl en la rama de RPV y de 10,4mg/dl en la rama de EFV (p<0,0001) y de triglicéridos de −6mg/dl en la rama de RPV y de 12,4mg/dl en la rama de EFV (p<0,0001); no hubo diferencias significativas en el cambio de la ratio de colesterol total/cHDL (−0,36 en la rama de RPV y de −0,28 en la rama de EFV; p=0,25). Las alteraciones de laboratorio grados 3–4 de incremento de colesterol total (el 0 frente al 3%), cLDL (el 1 frente al 6%) y triglicéridos (< 1 frente al 3%) fueron más frecuentes en la rama de EFV que en la de RPV. En el estudio ECHO5, al cabo de 48 semanas, los cambios fueron similares a los del estudio THRIVE, aunque menos marcados, probablemente debido a los análogos de nucleósidos acompañantes. En concreto, en el ensayo clínico ECHO el cambio medio de colesterol total fue de 1,1mg/dl en la rama de RPV y de 24,3mg/dl en la rama de EFV (p<0,0001), el cambio medio de cLDL fue de −1,5mg/dl en la rama de RPV y de 11,9mg/dl en la rama de EFV (p<0,0001), el cambio medio de cHDL fue de 2,7mg/dl en la rama de RPV y de 9,2mg/dl en la rama de EFV (p<0,0001) y el cambio medio de triglicéridos fue de −8,8mg/dl en la rama de RPV y de 14mg/dl en la rama de EFV (p=0,01); al igual que en el estudio THRIVE, no hubo diferencias significativas en el cambio de la ratio de colesterol total/cHDL (−0,14 en la rama de RPV y de −0,24 en la rama de EFV; p=0,25). En el estudio ECHO, las alteraciones de laboratorio grados 3–4 de incremento de colesterol total (el 0 frente al 2%), cLDL (el 1 frente al 2%) y triglicéridos (el 0 frente al 2%) fueron más frecuentes en la rama de EFV que en la de RPV, aunque menos marcadas en ambas ramas que las correspondientes del estudio THRIVE. El análisis conjunto de los estudios THRIVE y ECHO a las 96 semanas confirmó los cambios observados en ambos estudios por separado7 (fig. 4).
 de los lípidos desde el inicio.")
Más recientemente, en el estudio STaR10 se ha comunicado que al cabo de 48 semanas los cambios medios de los lípidos también fueron menores en la rama de RPV: colesterol total, 1 frente a 22mg/dl (p<0,001); cLDL, 1 frente a 14mg/dl (p<0,001); cHDL, 2 frente a 8mg/dl (p<0,001), y triglicéridos, −8 frente a 8mg/dl (p<0,001). Sin embargo, el cambio en la ratio colesterol total/cHDL fue idéntico en ambas ramas (−0,2).
En el ensayo clínico SPIRIT, la mayoría de los IP utilizados fueron atazanavir/r (37%), lopinavir/r (32,6%) y darunavir/r (20,2%). Tras 24 semanas11, todas las fracciones lipídicas disminuyeron significativamente en la rama de RPV siendo la media del cambio −25 frente a −1mg/dl (p<0,001) para el colesterol total, −16 frente a 0mg/dl para cLDL (p<0,001), −4 frente a −1mg/dl para cHDL (p<0,001) y −53 frente a 3mg/dl en el caso de los triglicéridos (p<0,001), siendo llamativo un descenso significativamente mayor de la ratio colesterol total/cHDL en la rama de RPV (−0,27 frente a 0,08; p<0,001). Estos cambios lipídicos son de dirección y magnitud similares a los observados con el cambio de IP potenciado con ritonavir a raltegravir en los estudios SPIRAL30 y SWITCHMRK31. En el primero de ellos, el cambio de IP por raltegravir se acompañó de un descenso significativo de marcadores de inflamación, resistencia a la insulina e hipercoagulabilidad32, quedando por dilucidar si este efecto también ocurre con la estrategia del estudio SPIRIT. Los datos del estudio SPIRIT a 48 semanas presentados recientemente confirman los resultados a 24 semanas11. Un estudio retrospectivo reciente ha evaluado en un centro de Londres la combinación de RPV/TDF/FTC en un solo comprimido33. A diferencia del estudio SPIRIT, el tratamiento previo contenía un INNTI (concretamente, EFV) en la mayoría de los pacientes. A pesar de esta diferencia, se observaron descensos en colesterol total, triglicéridos y en la ratio colesterol total/cHDL. Por tanto, la evidencia disponible muestra que el tratamiento con RPV tiene un escaso impacto sobre los lípidos y, en consecuencia, no contribuye a incrementar el riesgo cardiovascular de los pacientes que lo reciban.
Se ha comparado el efecto in vitro de RPV frente al de EFV en cultivos de adipocitos humanos12 observándose que EFV tiene un efecto antiadipogénico y proinflamatorio a dosis bajas, mientras que RPV a dosis comparables a las alcanzadas en pacientes tratados no tenía dichos efectos. Estos datos refuerzan el efecto neutro que tiene RPV sobre el metabolismo lipídico y el tejido graso.
En resumen, los datos de los diferentes ensayos clínicos sugieren que RPV se asocia a un excelente perfil de tolerabilidad, destacando una significativa menor tasa de efectos neuropsiquiátricos, comparado con EFV, una buena seguridad hepática y un perfil lipídico neutro. Todas estas características, unidas a la posibilidad de administrarlo en pautas sencillas, potenciarán en el futuro su utilización en el escenario de simplificación.
Conflicto de interesesLuis F. López Cortés ha recibido financiación para investigación, honorarios por consultoría, conferencias y reuniones de asesoría por parte de Abbott, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmith-Kline, Janssen-Cilag, S.A., Merck Sharp & Dohme, ViiV Healthcare y Roche Farma.
Esteban Martínez ha recibido financiación para investigación, honorarios por consultoría, conferencias, reuniones de asesoría o viajes a congresos por parte de Abbott, Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, GlaxoSmithKline, Merck Sharp & Dohme, Theratechnologies, Tibotec y ViiV Healthcare.
Miguel Ángel von Wichmann de Miguel ha participado en reuniones de asesoría y ha impartido conferencias en reuniones organizadas por Abbott, Bristol-Myers Squibb, Gilead Science, Janssen-Cilag, S.A., Merck Sharp & Dohme y Roche Farma.

