




La primoinfección por virus de Epstein Barr (VEB) se produce generalmente durante los primeros años de vida, aunque con la mejora de las condiciones económicas y sanitarias de las últimas décadas ha disminuido la tasa de infección en la infancia aumentando el número de adolescentes y adultos jóvenes susceptibles al virus1. El espectro clínico es amplio y depende de la edad de adquisición y de factores relacionados con el huésped. La infección es subclínica en la mayoría de los casos si se produce durante los primeros años de vida, pero si se retrasa hasta la adolescencia o juventud, se desarrolla mononucleosis infecciosa (MI) en el 25-75% de los infectados2. Aunque en individuos inmunocompetentes las manifestaciones clínicas suelen ser leves, recientemente se ha descrito el aumento de la incidencia de casos graves de MI relacionada con el VEB3.
Presentamos aquí los casos clínicos de dos hermanos que con un año de separación ingresaron en nuestro hospital por un cuadro de MI asociada a VEB atípico por la intensidad y la duración de los síntomas y por la aparición de complicaciones.
El primero de los casos corresponde a un chico de 14 años sin antecedentes patológicos previos y correctamente vacunado que ingresó en el hospital tras 20 días con fiebre alta persistente, astenia, anorexia, odinofagia y dolor costal derecho. En la exploración destacaba fiebre (38°C) y sudoración profusa, tinte ictérico en conjuntivas, taquicardia (114 lpm), eritema faríngeo con exudado, adenopatías cervicales y axilares, hipoventilación alveolar en ambas bases, hepatomegalia indolora (a 4-5cm del reborde costal) y esplenomegalia.
Presentaba leucocitosis en sangre periférica (66.200cel/μl) con un 67% de linfocitos que al microscopio tenían aspecto de linfocitos activados. Por citometría de flujo el 95% de la población linfocitaria presentaba marcadores de células T (CD 3, CD2, CD5 y CD 7) y un 5% presentaban marcadores de células B (CD19 y CD20). Entre los linfocitos T el 74% eran CD8+ y un 18% CD4+. Existía una hiperbilirrubinemia a expensas de la fracción directa (bilirrubina total 4,41mg/dl; directa 4,04mg/dl), elevación de transaminasas (GOT 241U/l; GPT 233U/l), láctico deshidrogenasa (LDH 2.259U/l) y un ligero alargamiento del tiempo de protrombina. En el estudio serológico se encontró un título alto (>1/256) de IgM contra el antígeno de la cápside de VEB (MeriFluor EBV VCA IgM, Meridian Bioscience, Inc. EE.UU.). El estudio serológico frente a CMV, VIH, VHB y VHC fue negativo. En la radiografía de tórax se demostró derrame pleural en el hemitórax derecho y el líquido extraído mediante toracocentesis cumplía criterios de Ligh para exudado, incluyendo 3.520 leucocitos/mcl (76% polimorfonucleares y 24% mononucleares). El ADN fue extraído del líquido pleural usando un kit comercial (High Pure Viral Nucleic Acid Kit. Roche, Barcelona, España) y amplificado mediante una PCR diseñada para detectar varios virus de la familia Herpesviridae. En la electroforesis sobre gel de agarosa se detectó una banda de ADN de 535pb, este ADN era digerido por un enzima de restricción específica para el VEB4. La ecografía abdominal confirmó la hepatomegalia difusa y la presencia de ascitis en cuantía moderada. Se inició tratamiento con metilprednisolona (0,8mg por kilogramo de peso) después de lo cual comenzó una progresiva mejoría sintomática. El paciente fue dado de alta después de 14 días, pero el glucocorticoide se administró durante 35 días y se retiró progresivamente.
Un año más tarde ingresó el hermano mayor del primer paciente, un varón de 19 años de edad. Era estudiante universitario y tres semanas antes del ingreso había comenzado con fiebre, adenopatías laterocervicales bilaterales, rash cutáneo y dolor faríngeo. Había sido seguido de forma ambulatoria, pero se decidió su ingreso ante la intensa afectación del estado general.
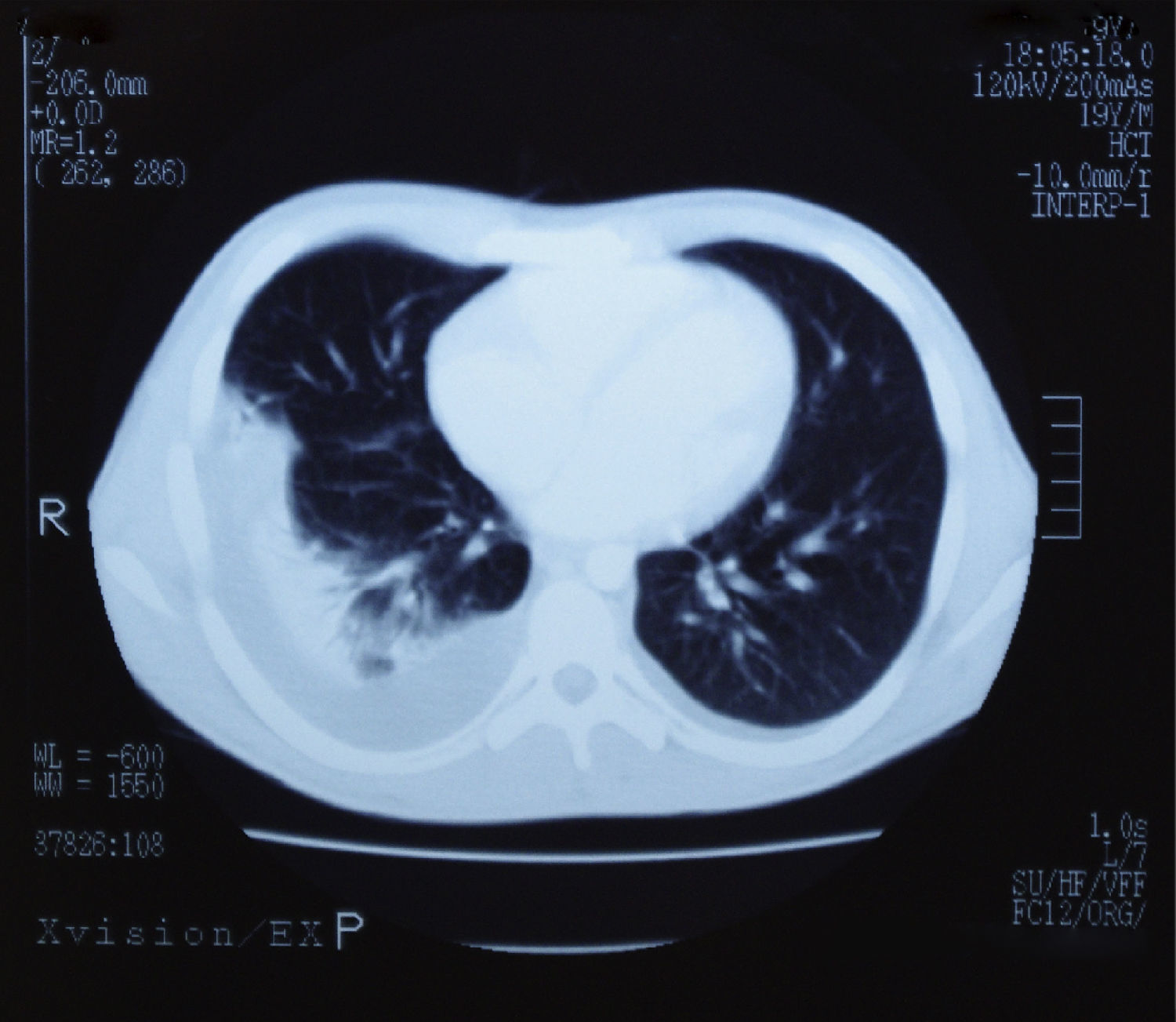
Presentaba también leucocitosis (24.500cel/μl) con predominio de linfocitos (71%) que en el frotis tenían aspecto atípico en un alto porcentaje, trombopenia ligera (106.000plaquetas/μl), alargamiento discreto de los tiempos de coagulación (INR 1,7) y datos de colostasis (bilirrubina directa 3,1mg/dl). Tenía un título alto (>1/256) de IgM frente al antígeno de la cápside del VEB. El estudio serológico de CMV, VHA, VHB y VIH era negativo. Una TAC de tórax y abdomen puso de manifiesto la presencia de esplenomegalia, hepatomegalia, ascitis y derrame pleural bilateral (fig. 1). Se instauró tratamiento con metilprednisolona (1mg/kg/día) que se mantuvo durante 30 días. Tras su inicio remitió la fiebre y se produjo una lenta, pero progresiva mejoría en un periodo de 35 días. El paciente fue dado de alta a los 10 días, pero en el seguimiento posterior refería debilidad y dificultad para la concentración durante tres meses más. Los parámetros hepáticos se normalizaron a los 6 meses del inicio de los síntomas.

Después de dos y un año de seguimiento respectivamente, ninguno de los dos pacientes presentó datos de síndrome linfoproliferativo, gammapatía monoclonal ni inmunodepresión.
Se trata pues de dos casos confirmados de MI grave asociada al VEB con manifestaciones poco frecuentes; de hecho, aunque la infección por VEB puede afectar en teoría a cualquier órgano, el derrame pleural y la ascitis son manifestaciones escasamente descritas en la literatura5. La intensidad y la duración de los síntomas son también inusuales en estos dos casos; en un estudio reciente que incluyó a 58 universitarios de Reino Unido con MI asociada a VEB la duración media de la sintomatología aguda fue de 7 días2 mientras que en nuestros pacientes la sintomatología de la fase aguda se prolongó aproximadamente 35 días y la normalización bioquímica unos 10 meses después en el primer caso. En ese mismo estudio el recuento medio de linfocitos fue de 5.800 frente a la elevada linfocitosis de nuestros casos (44.350 y 17.395cel/μl).
El segundo de los casos nos hizo plantearnos la posibilidad de que existiera un factor familiar que determinara la susceptibilidad a una infección severa por VEB. Existen diversas publicaciones que relacionan determinados aspectos genéticos del huésped con la infección por VEB6–9, a continuación describimos someramente los más conocidos. El síndrome linfoproliferativo asociado al X (X-linked lymphoproliferative síndrome; XLS) también conocido como síndrome de Duncan es una inmunodeficiencia congénita que afecta a 1 de cada 106 varones. Los pacientes con este trastorno son normales hasta que ocurre la primoinfección por el VEB. La manifestación más frecuente es una mononucleosis severa o fatal en algunos casos que puede ser idéntica a un síndrome hemofagocítico asociado a virus. Los trastornos linfoproliferativos, las disgammaglobulinemias (hipogammaglobulinemia) y otras alteraciones hematológicas (anemia aplástica) son otras de las manifestaciones de este síndrome. El gen responsable del X-linked lymphoproliferative sindrome ha sido identificado (SH2D1A); este codifica una proteína de 128 aminoácidos que juega un importante papel en las vías de transducción de señales en los linfocitos T. La mutación de este gen produce una proliferación incontrolada de los linfocitos T CD8+1,6.
Estudios recientes también han puesto de manifiesto la asociación que existe entre ciertos alelos del sistema HLA (HLA-A*01) con el desarrollo de MI y de linfoma de Hodking asociado a VEB, de manera que las células con expresión restringida de HLA A01 fracasaban en la respuesta al VEB7.
Dada la trascendencia clínica que podría tener para estos dos hermanos remitimos muestras de sangre del segundo caso al centro de referencia para tipado de los antígenos del sistema HLA y a un laboratorio de investigación oncológica para estudio de posibles mutaciones en el gen SH2D1A. Para el HLA se realizó un tipado de baja resolución de los loci HLA A*, B* y DRB1* usando un sistema de PCR (SSP) (AllSet™ Gold HLA-A, HLA-B and HLA-DR Low Resolution SSP kits, Invitrogen Ltd, Paisley, Reino Unido) siendo el tipado: A*11/-B*35/-DRB1*03/11. No se detectaron mutaciones (cambio de aminoácidos, deleciones o inserciones) del gen SH2D1A usando PCR.
Aunque no hemos encontrado la base genética que pueda explicarla, pensamos que puede existir una predisposición de naturaleza genética que favoreció el desarrollo de estos dos casos graves de mononucleosis. El seguimiento en el futuro permitirá comprobar si dicho fenómeno tiene consecuencias a largo plazo.
Al Dr. Felipe Fernández Cuenca de la Unidad de Enfermedades Infecciosas y Microbiología Clínica del Hospital Virgen Macarena y a la Dra. María Francisca González Escribano del Servicio de Inmunología del Hospital Virgen del Rocío de Sevilla. Financiado por el Ministerio de Ciencia e Innovación, Instituto de Salud Carlos III, cofinanciada por el Fondo Europeo de Desarrollo Regional FEDER, Red Española de Investigación en Patología Infecciosa (REIPI RD06/0008).

