




Introduction
The acquired immune deficiency syndrome is currently one of the main causes of mortality in the world. Antiretroviral therapy reduces viral replication and disease progression, with the result that mortality has fallen in countries which have access to therapy. Nevertheless, currently available drugs cannot eradicate the virus and therapy is considered as being "for life". Prolonged treatments and their difficult follow-up (poor adhesion), have brought to light problems which were originally expected such as resistance to antiretrovirals, unexpected adverse effects (e.g. mitochondrial toxicity), redistribution of body fat and metabolic alterations which can lead to secondary diseases. The prevalence of primary or acquired resistance to available drugs is increasing. Therefore, it is essential to find new drugs which can act on known targets (reverse transcriptase and protease), as well as others which act on new targets, with minimum toxicity for the host in both cases1,2.
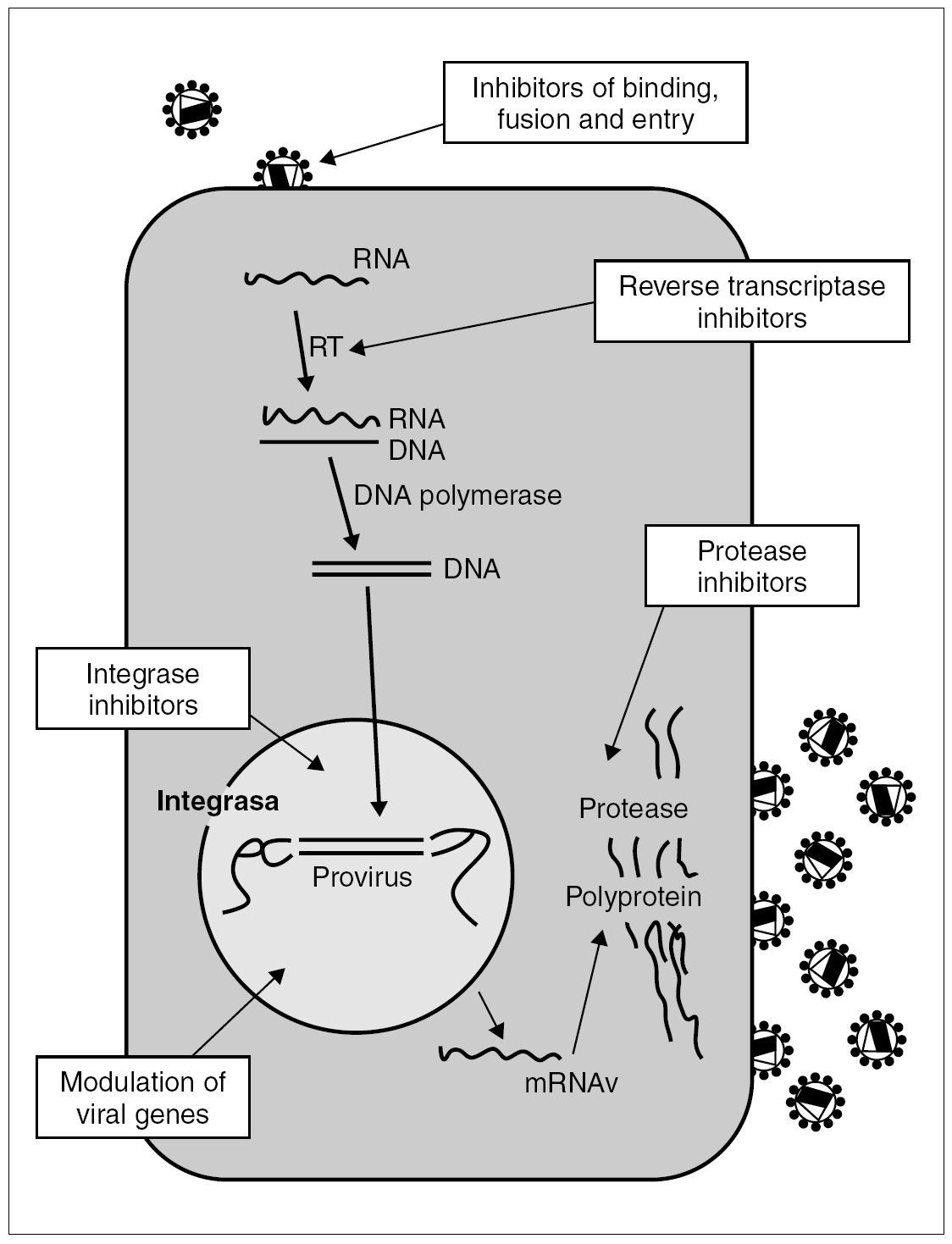
The most promising therapeutic targets are the process of viral binding and penetration of the cell and the integration of viral DNA into the host genome. In the longer term, we can envisage other targets, such as action on the viral genome and the use of host cell biological mechanisms to inhibit viral replication (fig. 1).

Figure 1. HIV vital cycle. Antiretroviral drug targets.
Drugs which act on new targets
HIV binding and penetration of the cell
The human immunodeficiency virus (HIV) is covered by a double-lipid layer speckled with viral glycoproteins of which the most important are gp120 and gp41. These are related and stem from a larger common precursor: gp160. gp120 stems from the terminal amino portion of gp160 and is situated at the most external part of the viral lipid layer; this glycoprotein intervenes in the binding of the CD4 receptor and "directs" the fusion mechanism. gp41 stems from the C-terminal portion of gp160; it is a transmembrane protein and plays an important role in membrane fusion. The infectivity of a virus requires the presence of both glycoproteins3.
The amino acid sequence of gp120 is formed by five variable regions (V1 to V5), which alternate with more conserved regions. The variable regions are the most exposed of the viral surface. The binding site of the virus to the CD4 receptor is formed by specific regions of gp120 which use the primary sequence and their spatial conformation to create a domain which is capable of recognizing its target (CD4 receptor) and binding to this with greater of lesser affinity according to the existence or not of mutations in glycoprotein gp120 or in the CD4 receptor3,4.
From the pathophysiological viewpoint, we can consider three phases, or mechanisms, of viral entry to the cell4-6 and each can be a therapeutic target: a) adhesion of the virus to the CD4 receptor, b) binding of chemokines to the receptors which act as HIV co-receptors, and c) fusion of viral and cell membranes.
Binding of the virus to the CD4 receptor
This step is mediated by gp120 and the domain which recognizes the CD4 receptor. The appearance of gp120 is that of a spike (trimer) formed by different loops (V1-V5) and in which the binding region has been described in the conserved part of the V1-V2 trunk and near V3. Some biological activities of the virus such as cellular tropism, pathogenicity, capacity for fusion and use of co-receptors, seem to be mediated by a domain in the V3 loop. After binding of gp120 to the CD4 receptor, conformational changes are introduced which bring the virus closer to the chemokine receptors.
Binding to co-receptors
Changes in the conformation of gp120 reveal the epitopes required for binding to chemokine receptors. Chemokines are small proteins with a pro-inflammatory function released by macrophages, activated T lymphocytes and other mononucleated cells (in the case of MIP-1α, MIP-1β and RANTES) or produced by stromal cells (in the case of SDF-1) which, when bound to their receptors, transmit a signal to the inside of the cell by activating chemoattraction. Most chemokines can be grouped according to a sequence of amino acids which is characteristic in two families: CC and CXC. The production of chemokines and the availability of their receptors depend on several factors, such as the presence of cytokines, infections by microorganisms, etc.7,8.
The chemokine receptors used by retroviruses to enter the cell are the true HIV receptors, as some genetically modified HIV strains have been observed to be capable of entering the cell without using the CD4 receptor9. The CCR5 receptor of the β-chemokines has been identified as a co-receptor of the recently infected viruses, with a tropism for macrophages with this receptor in their membrane and with little capacity to form syncytia in lymphoid cell lines. The virus used by this receptor is known as HIV R5. The importance of the CCR5 receptor during the initial moments of infection can be evaluated by the fact that people lacking it (those who are homozygotic for the deletion of 32 base pairs in the CCR5 gene known as polymorphism Δ -32) are resistant to infection by HIV R5, and in heterozygotes, the infection progresses more slowly. Nevertheless, these people can be infected by the CXCR4 receptor, although this is very rare. Furthermore, viruses using the CXCR4 co-receptor (virus R4) infect both macrophages and lymphocytes, are more common in advanced stages of infection and have a high cytolytic and syncytia-forming capacity.
Fusion of the cell and viral membranes
Fusion of both membranes is the last step in the mechanism of viral entry into the cell and is mediated by gp41. This is a transmembrane glycoprotein whose most distal portion, the aminoterminal portion, contains a glycine-rich hydrophobic radical (where the fusion peptide is formed) which is essential for binding of the membranes. Between the N-terminal and C-terminal portions of gp41 there are two helicoidal structures, or heptad repeats (HR), each identified as HR-1 and HR-2. HR-1 is distal to the virus and closer to the fusion peptide. In the free virion, gp41 has a non-fusogenic conformation, but when gp120 binds to its receptor, gp41 undergoes a conformational change and an elongation (pre-intermediate hairpin) is formed which inserts the fusion peptide into the target cell membrane. After insertion, HR-2 folds into three helicoids on the three HR-1 helicoids, forming a six-helix structure whose objective is to further reduce the distance between the virus and the cell until it brings both membranes into contact so that they finish by merging.
Blocking entry of the virus
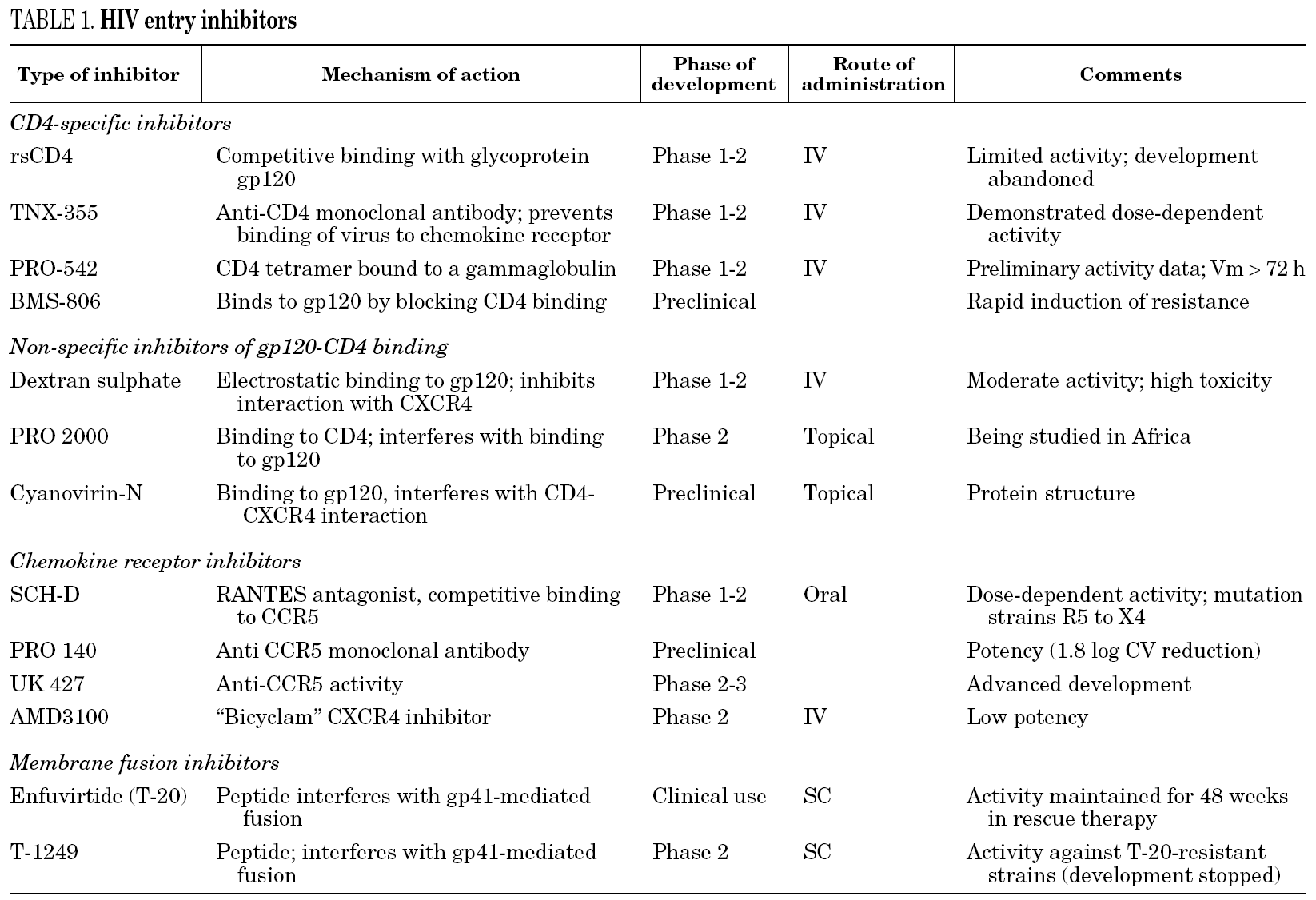
The drugs which act on any of three steps mentioned above can block entry of the virus to the cell. Its targets can be any of the two glycoproteins of the virus (gp120 and gp41) or their corresponding cellular receptors or co-receptors. The number of substances with this capacity now being studied is very high, most are in early phases of investigation and very few are likely to be used in clinical practice (table 1). Nevertheless, a drug is available (T-20, enfuvirtide or Fuzeon®) whose role as a component of antiretroviral therapy is being defined.

Inhibitors of HIV binding to CD4
Non-specific inhibitors
Polyanionic molecules. Certain sulphated polysaccharides (dextran-sulphate, pentosan-sulphate or heparin) can inhibit viral replication in vitro. The anti-HIV activity of compounds with structures which are so heterogeneous seems to be due to the fact that they have a high density of negative charges (polyanions). Dextran-sulphate binds to the V3 loop of gp120 in CXCR4 strains and prevents binding to the CD4 receptor. Other substances which share this mechanism of action are PRO 2000 and cyanovirin-N. Although they are unlikely to be used in clinical practice, different polyanions are currently being evaluated as topical agents6.
Specific inhibitors
Soluble recombinant CD4 (srCD4). srCD4 was one of the first antiretroviral drugs to be studied. It acts by blocking gp120 and has proven to very active in vitro. Nevertheless, its activity is much lower against viral strains from patients, possibly because there are viral variants of lower affinity. Therefore, srCD4 has not been developed as an anti-HIV agent5,6.
Anti-CD4 monoclonal antibodies. These are monoclonal antibodies anti determined epitopes of the CD4 receptor or of gp120 which, on binding, block interaction and repress the replication of several viral sub-types. Even though blockade of the CD4 receptors could lead to some degree immunosuppression, at least the so-called TNX-355 antibody has been well tolerated and CD4+ lymphocytopenia has not been reported5. A first clinical trial (phase I/II) at a single dose of TNX-355 showed a reduction in viral load and an increase in CD4+ cells at 21 days10. Nevertheless, longer clinical trials are necessary to confirm the usefulness of this antibody. PRO-542 is a hybrid tetramer which contains domains of the CD4 receptor bound to IgG2 and which acts as a decoy of the CD4 receptor to which viral gp120 binds, thus preventing it from binding to the real CD4 receptor. In vitro, it has shown activity with both laboratory and clinical strains, and this activity has been confirmed in vivo in a small number of advanced-stage patients11.
BMS-806. This substance forms part of other molecules which can bind competitively and reversibly to gp120 by blocking the interaction with the CD4 receptor. It has been evaluated in vitro and is active against different strains of HIV, although the early emergence of mutation-related resistance at the point of action of gp120 leads it to lose efficacy12,13.
Substances which block binding to chemokine receptors
The discovery of co-receptors for entry of the virus has opened up new possibilities in the development of anti-HIV drugs. Attempts have been made to emulate the structure of the natural cytokines which block these receptors and inhibit viral replication14,15. Early-stage studies are being carried out on drugs which block one or the other co-receptor, with doubts as to whether blockade can induce mutations in the strain, thus making it easy for the virus to use the other receptor.
CCR5 receptor antagonists5
Tak-220. Pre-clinical studies of this molecule have shown high specificity for CCR5 with no affinity for other ligands. Similarly, oral administration enables suitable levels of the drug in blood to be reached.
SCH-C/SCH-D. These are substances have a high intrinsic activity against R5 strains and are synergic with other antiretroviral drugs. SCH-C has undergone a phase I/II trial in 12 patients for 10 days and has shown a reduction in viral load of between 0.5 and 1.0 log. Nevertheless, its development has been stopped because of the risk of cardiac arrhythmias after reports of a lengthening of the QTc interval16,17. It has been replaced by SCH-D, which has recently been reported to be more potent (mean reduction in viral load of 1.3 log10), is better tolerated and has no effect on heart rate18. Nevertheless, it is important to note that one of the 48 patients studied developed a mixed viral strain, R5/X4 and another developed an X4 strain on finishing therapy.
UK-427,857. This drug is active against a wide number of viral strains and is specific for the CCR5 receptor (not active against CXCR4 strains). It is currently in phase II/III studies.
Other substances. Other substances in early stages are Pro-140, which is a specific and potent monoclonal antibody (reduction in viral load to 1.8 log), can inhibit entry of the virus and does not block CCR4 receptor activity. GW873140 has shown good tolerance (digestive symptoms) and a prolonged half-life after oral adminstration19. AMD887 is active and potent in vitro, both as the only drug and in combination with AMD070 (CXCR4 co-receptor inhibitor)20.
CXCR4 receptor antagonists5
Bicyclams. These molecules are so named because they are composed of two macrocyclical rings (cyclams) bound by an aliphatic or aromatic chain. They are very potent and are active against HIV-1 and HIV-2 in cell culture, but have been ruled out as drugs in clinical trials. More information is available for AMD3100. In vitro studies in which peripheral mononucleated cells infected with strains X4 and R5 were placed into contact with AMD3100 revealed blockade of the CXCR4 receptor and they were only collected in the R5 virus supernatant. In another study with peripheral blood lymphocytes infected by syncytia-forming viruses, these strains lost their capacity to form syncytia by mutating to use the CCR5 co-receptor21. This mutation can shorten the course of the disease. Nevertheless, the development of AMD3100 has been stopped because of the adverse effects observed in a phase I study, although other compounds, such as AMD070, are being studied, and have shown in vitro activity against a large number of strains which are resistant to several currently used drugs.
KRH-1636 and KRH-2731 are potent antagonists of the CXCR4 receptor, can be administered orally and are active in pre-clinical studies22,23.
Blockade of viral fusion with the cell membrane
Substances which are currently known to block fusion of the viral capsid and cell membrane are synthetic peptides which camouflage the HR-2 region of gp415. They act on the attack hairpin by inhibiting its formation. This attack hairpin is necessary for fusion of the virus. One of the most important of these substances is enfuvirtide (T-20) and T-1249.
Enfuvirtide is a 36-aminoacid peptide which camouflages a sequence of the HR-2 region and is active against strains X4 and R5. It binds to the HR-1 domain of the gp41 virus by preventing the formation of the 6-helix structure necessary to start the conformational changes which finish in membrane fusion. The drug is active during the phase where the virus approaches the target cell in which gp41, and specifically the HR-1 domain, are accessible. This "therapeutic window" can vary according to the affinity of the virus for the receptor, in such a way that, if affinity is very high, the drug can act more quickly and its efficacy is lower in these viral strains5. The efficacy of enfuvirtide is not modified independently of the co-receptor used by the virus24.
Enfuvirtide is marketed in many countries. Initial studies using the drug in intravenous monotherapy showed a potent antiretroviral effect with few adverse effects. This activity was proven both for sub-type B (dominant in Europe and the USA) and against other viral sub-types. The optimal subcutaneous dose was initially determined in a continuous perfusion pump and later in two daily doses in 16 adults who achieved a negative viral load (< 500 cop/ml) when the dose was above 100 mg. Studies in vitro have shown the synergy of enfuvirtide with other binding blockers such as AMD-3100, SCH-C and PRO-54225.
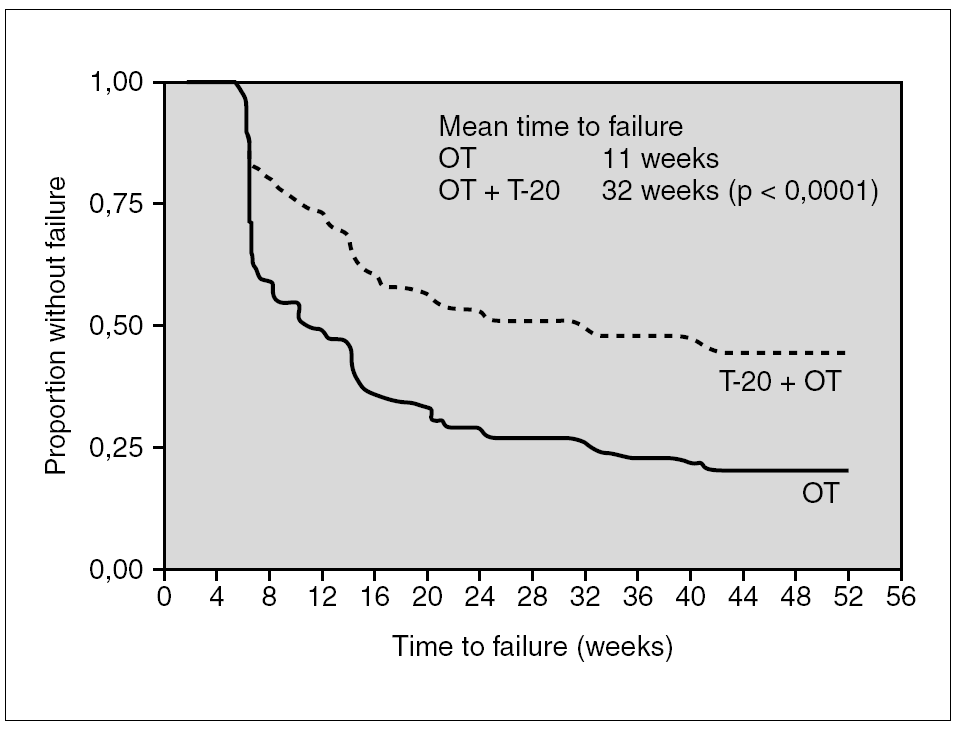
The results of two parallel studies (TORO-1 and TORO-2) which included 995 very experienced patients have been essential in allowing health authorities to approve this drug in clinical practice. These trials compared the response to the best treatment possible (three to five drugs) determined using genotypic and phenotypic resistance studies, by adding enfuvirtide to a group of the study subjects (proportion 2:1). At 24 and 48 weeks, both the reduction in viral load and the increase in CD4+ lymphocytes were higher in the group which combined with enfuvirtide. The median time to virological failure (taking into account that the trial involved multi-treated patients) improved from 11 to 32 weeks26-28 (fig. 2).

Figure 2. Time to protocol defined virologist failure in patients with three drug families' resistance and optimized treatment (OT) with or without enfuvirtide (OT + T-20).
The adverse effects were similar in both groups except for local reactions at the injection point. Global incidence of bacterial disease was similar in both groups, but in the enfuvirtide treated group, a greater incidence of pneumonia was observed, with no apparent cause. As far as laboratory alterations were concerned, there were reports of an increase in peripheral eosinophilia in the enfuvirtide arm, although cases of hypersensitivity are rare. The injection site reaction involved painful inflammatory nodules which ocurred in most patients and lasted from one to three days. Only a small number of patients modified their daily activities or needed pain killers for the nodules, although 2.8% stopped treatment for this reason29.
The efficacy of enfuvirtide in monotherapy is transitory, therefore it must be combined with other antiretrovirals to which the virus is susceptible. Resistance to enfuvirtide is related to mutations in the gp41 gene. The HR-1 and HR-2 domains are relatively stable, but the reported cases of resistance are related to mutations in codons 36 to 45 of gp41 with a variable reduction in susceptibility of between 9.1 and 45 times. The viruses which present this mutation have reduced fitness25.
Enfuvirtide is indicated in HIV+ patients who have failed with regimens of at least one drug from the three groups of antiretrovirals or who have developed intolerance to previous therapy. Given that the TORO studies showed a better virological response if enfuvirtide is combined with active drugs, the drug must be administered before patients have run out of therapeutic options.
T-1249. This is a second-generation fusion inhibitor. It is a 39-aminoacid peptide designed from different HR-2 regions of HIV-1, HIV-2 and SIV. As with enfuvirtide, T-1249 has a high capacity for reducing viral replication in HIV strains which are multi-resistant to current drugs, even those which are resistant to T-20, and in sub-types A to G. In naïve patients and those on monotherapy, T-1949 has proven to be very potent at a single daily dose (fall in viral load of 2.0 log)5,30. Nevertheless, the clinical development program of this product has been temporarily stopped because of technical difficulties.
Other, less complex peptides are being designed which act on the most distal portion (amino-terminal) of gp41 near the fusion peptide31, although the current challenge is to obtain drugs which are able to block these steps in the viral cycle, but which are administered orally in such a way as to simplify the treatment of this chronic disease.
Integration of proviral DNA (Integrase inhibitors)
The integration of proviral DNA in the host chromosome is a necessary step in the HIV replication cycle and integrase is the key integration enzyme. This enzyme is encoded in the HIV genome together with reverse transcriptase and protease. Integrase is a very therapeutic target since, in addition to being crucial for viral replication, there is no enzyme in human cells which carries out these functions, and therefore, drugs which block it should not affect other metabolic processes32.
Integrase is a 32-kD protein which processes and transports viral DNA to the interior of the nucleus and catalyzes its insertion into the DNA of the host cell using two sequential reactions. It eliminates two nucleotides of each 3´ terminal portion of the viral DNA (3´ processing) before integrating viral DNA into the genome of the host cell using trans-esterification (transfer of the DNA chain). Both integrase itself and the so-called integration complex (integrase-viral DNA) could be targets for pharmacological action33.
Despite the fact that the active structure of integrase has been known since the beginning of the last decade, no drug is available at present. We are aware of a wide variety of chemical substances with the ability to block integrase in vitro (oligonucleotides, curcumin analogs, polyhydroxylated aromatic complexes, di-ketoacids, caffeoyl- and galloyl - based compounds, hydrazides and amides, tetracyclines, etc.), although, unfortunately, they do not display this activity at the cell level34,35. Nevertheless, a further step has recently been taken with the report of the "catalytic domain-inhibitor" complex, which is giving new life to the study of chemical structures such as di-ketoacids and tartaric di-caffeoyl acids36.
The two most widely studied structures are V-165, which has proven to be potent in vitro and is active against strains resistant to reverse transcriptase and protease inhibitors, and synergic with some currently marketed antiretrovirals, and S-1360, a di-ketoacid in phase I studies with very similar tolerance and efficacy characteristics.
Integrase can undergo mutations in its active locus. Nevertheless, the susceptibility of the virus to products currently being studied does not seem to be reduced37.
Other possible targets
In addition to the targets mentioned above, we can envisage other possible therapies which are currently a long way off. These involve the mechanisms which condition viral latency, action on genes or the first steps in the synthesis of viral proteins.
One factor which appears to have an influence on the efficacy of viral replication is the glycoprotein APOBEC3G (apolipoprotein B, mRNA editor enzyme, polypeptide-like catalytic 3G, or CEM15). APOBEC3G is an innate intracellular antiretroviral factor which is countered by the lentiviral vif gene protein and which is species-specific. APOBEC3G acts as a cytosine deaminase and, in the viral DNA chain, it changes cytosine for uracil during retrotranscription, thus producing a hypermutation which makes the virions of following generation unviable. Nevertheless, the HIV-1 vif gene is capable of blocking APOBEC3G by avoiding its incorporation into offspring virions, thus facilitating its degradation in the proteasome38,39.
New drugs with action against known targets
The search for new drugs aims to increase potency, modify resistance profiles, reduce toxicity and simplify administration. Drugs with these characteristics which can soon form part of the armamentarium are the nucleoside analog (NA) emtricitabine, the non-nucleoside analog (NN) caprarvirine and the protease inhibitors (PI) atazanavir, fosamprenavir and tipranavir.
Emtricitabine (FTC) is a cytidine analog which is administered once daily (200 mg). Its resistance profile is similar to that of lamivudine, but its tolerance is 4-10 times higher, with the result that the mutation M184V, which generates high-grade resistance, appears less frequently. This drug is already available in some countries, given that it displays similar or greater efficacy to lamivudine and stavudine, and its minimal mitochondrial toxicity make it ideal for combining with tenofovir40.
Amdoxovir (DAPD) is a guanosine analog with activity similar to that of ABC, 3TC or d4T and an antiviral effect against the hepatitis B virus. It is active against strains which are resistant to ZDV, 3TC, ddI, ddC and ABC. Phase I/II studies are currently being carried out among experienced patients41, although its future remains unclear as development has been stopped temporarily.
Capravirine is a potent NN against viral strains resistant to other NNs. At least two simultaneous mutations (L100I, K103N, V106A) are necessary for resistance to appear. It is currently undergoing phase II testing in NN-experienced patients42.
TMC-125 is a new NN which, in vitro, shows equal potency in HIV wild-type and HIV strains which are multi-resistant to NNs (strains with the mutations L100I, K103N, Y181C, Y188L and G190A). In phase II studies of patients with at least two resistance mutations to NVP and EFV, an acceptable response and good tolerance have been observed43.
Atazanavir is a protease-inhibiting azapeptide whose pharmacokinetics allows once-daily administration. As well as being administered in a single dose, it requires fewer tablets than other drugs (two per day, or three if boosted with ritonavir). It has shown a potency similar to that of efavirenz and in naive patients in their first failure with PI, it has proven to be not inferior to lopinavir/ritonavir (in this case, atazanavir was boosted with ritonavir) at one year of treatment, although the number of mutations to PI at the beginning of the study is unknown44. It presents a favorable safety profile, and does not alter either plasma lipids or hydrocarbonate metabolism, although it does produce indirect hyperbilirubinemia in 10-33% of patients. Furthermore, the resistance profile of ATV seems to be totally different from that of other PI. Mutation I50L, which is specific for resistance to ATV, is different to the I50V observed in patients treated with amprenavir. The mutation I50L considerably reduces sensitivity to ATV, but increases sensitivity to other PIs45-47.
Fosamprenavir is an amprenavir prodrug. With this presentation, the number of tablets to be taken is reduced considerably and, when boosted with ritonavir, it can be administered once daily. The efficacy of the drug has been proven in naive patients ("Neat" study with BID dosing and "Solo" study with QD dosing) in whom it was compared with nelfinavir, and in both studies the response was similar to that of nelfinavir. In experienced patients, it has been compared with lopinavir/ritonavir and the response measured as the number of patients who reach an undetectable viral load or maintain an undetectable viral load for 48 weeks ("Context" study) somewhat inferior to that of the comparator. Nevertheless, it has been approved in some countries48,49.
Tipranavir is a non-peptidic PI which has shown activity in vitro against viral strains which are resistant to current PI50. In patients presenting their first failure with PI who were randomized to receive NA with tipranavir (500 or 1200 mg)/ritonavir (100 mg) or saquinavir (400 mg)/ritonavir (400 mg) BID, the fall in VL was 1.4 and 1.8 log, at 16 weeks. In patients presenting their second failure to PI treated with tipranavir (500 or 1000 mg)/ ritonavir (100 mg) every 12 hours, the fall in viral load at 48 weeks was 2.4 log in the group with five or fewer PI mutations, and 2.2 log in the group with more than five mutations50-52.
TMC-114 is a non-peptidic PI in the early stages of investigation. In vitro, it has been highly active against viral strains with high-level resistance to current PIs. In healthy volunteers, doses over 800 mg have been shown to reach blood levels equivalent to the CI50 of multi-resistant strains. It can be pharmacokinetically boosted with ritonavir and its most marked adverse effects are gastro-intestinal, headache or dizziness53,54.

