




El lopinavir/ritonavir (LPV/r) ha demostrado eficacia virológica e inmunológica en el tratamiento antirretroviral (TAR) combinado, tanto en pacientes naïve como pretratados. Además presenta una alta barrera genética al desarrollo de resistencias y su perfil de tolerancia es aceptable, aunque son frecuentes alteraciones gastrointestinales y del perfil lipídico.
Se revisan diferentes estrategias utilizadas en la optimización del TAR con este fármaco en la práctica clínica diaria, haciendo especial incidencia en la monitorización de concentraciones plasmáticas de LPV/r y la caracterización farmacogenética de las principales isoenzimas responsables de su metabolismo y transporte. En este sentido, la correlación del genotipo con el fenotipo establecida en la monitorización de niveles de LPV/r facilitaría la individualización posológica de los tratamientos con este fármaco. Así mismo, se revisa la estrategia de simplificación del tratamiento a monoterapia, lo que permitiría incrementar la seguridad y disminuir los costes.
Lopinavir/ritonavir (LPV/r) has demonstrated virological and immunological efficacy in the combined antiretroviral treatment (cART), in both naïve and experienced patients. Furthermore, LPV/r showed a high barrier to the development of resistance. Although generally well tolerated, adverse gastrointestinal side effects and metabolic disorders are frequent.
The different tools used to optimise the cART with this drug combination in the daily clinical practice, emphasising the therapeutic drug monitoring (TDM) of LPV/r and the genetic analysis of the main enzymes responsible for the metabolism and transport, are reviewed. The relationship between phenotype and genotype, established through TDM, could be useful for the physician to improve the clinical management of the HIV infection, due to the possibility of individualising the dose with this drug. Monotherapy is also reviewed as a new strategy used in the simplification of the treatment with this drug, which could increase safety and reduce costs.
El síndrome de inmunodeficiencia adquirida continúa siendo una prioridad sanitaria mundial, ya que es una de las primeras causas de muerte en los países en vías de desarrollo1. Aunque los efectos beneficiosos del tratamiento antirretroviral (TAR) combinado son indiscutibles, con frecuencia los acontecimientos adversos asociados a la terapia farmacológica pueden comprometer la calidad de vida y afectar la adherencia.
La introducción de los inhibidores de la proteasa (IP) a mediados de la década de los noventa como tercer componente del TAR mejoró radicalmente los resultados del tratamiento de la infección por el virus de la inmunodeficiencia humana (VIH). La aparición del lopinavir (LPV) como el primer IP coformulado con el potenciador ritonavir (RTV) en el año 2001 constituyó uno de los grandes avances en la historia del TAR2,3, lo que ha contribuido de manera importante a que el LPV potenciado con RTV (LPV/r) desplazara al resto de IP de primera generación4.
En el momento actual, aunque se continúa trabajando en el diseño y el desarrollo de nuevos fármacos dirigidos a otras dianas del VIH, también existen investigaciones paralelas enfocadas hacia la optimización de la terapia con el arsenal terapéutico actualmente disponible. La farmacocinética (PK) y la farmacogenética (PG) son disciplinas que paulatinamente están demostrando su utilidad en la optimización del TAR, especialmente en los centros hospitalarios que cuentan con la tecnología necesaria para su aplicación en la práctica clínica habitual. De hecho, existen documentos de consenso y publicaciones científicas que avalan la utilidad de la monitorización de niveles de fármacos (therapeutic drug monitoring, TDM) antirretrovirales (ART) en ciertos escenarios terapéuticos5–11 y de algunas determinaciones genéticas, como por ejemplo el HLA B*5701, CYP2B6*6 y SLCO1B1*4, para el tratamiento con abacavir5–9, efavirenz10–14 y LPV/r15, respectivamente. En este documento se pretende revisar la información disponible sobre la utilización de estas herramientas PK y PG aplicadas a LPV/r, así como las nuevas estrategias en los esquemas de tratamiento enfocados hacia la utilización de LPV/r en monoterapia16 y a la administración de una única dosis diaria17.
Propiedades farmacodinámicasLa proteasa del VIH es una proteína compuesta por 99 aminoácidos y es responsable de la maduración de las partículas del virus. En el VIH se originan tres grandes precursores codificados por los genes: env, gag y pol. Las poliproteínas resultantes gag y pol son procesadas por la proteasa del VIH, que cataliza la escisión de estos precursores polipeptídicos en subunidades funcionales para la formación de la cápside viral y de las enzimas virales, necesarias para dar un virión infeccioso18. Los IP actúan como inhibidores competitivos que se unen directamente a la proteasa del VIH-1 y VIH-2, bloqueando así la escisión de las poliproteínas gag y pol, previniendo la ruptura posterior de los polipéptidos y por tanto la maduración del virus, y dando lugar a viriones no infecciosos18. Aunque todos los IP tienen este mecanismo de acción, presentan entre ellos importantes diferencias en la eficacia y en el perfil de eventos o acontecimientos adversos5,7.
Propiedades farmacocinéticasEl comportamiento PK de LPV/r condiciona las concentraciones que alcanza este fármaco en los diferentes órganos y tejidos, así como su permanencia en el organismo. Considerando que las concentraciones de fármaco constituyen una variable subrogada de la respuesta, resulta de gran interés conocer sus características PK.
La absorción de LPV/r tras su administración en forma de cápsulas blandas o solución, formulaciones inicialmente comercializadas (Kaletra®, Laboratorios Abbott), estaba claramente influida por la presencia de alimentos. Así, tras la administración de una dosis única de 400/100mg en cápsulas blandas con una dieta de moderado contenido graso, la concentración máxima (Cmax) y el área bajo la curva (ABC) de LPV se incrementaron un 23 y un 48%, respectivamente, y un 54 y un 80% para la solución oral2, con respecto a la administración en ayuno. Por ello se aconseja su administración con las comidas con el fin de mejorar su biodisponibilidad, que presenta una elevada variabilidad intra e interindividual. Con objeto de evitar estos problemas, en 2006 se aprobó una nueva formulación en comprimidos con la tecnología de melt-extrusion, que ha mostrado una exposición a ambos fármacos similar, independientemente de la presencia o no de alimentos, y una absorción más rápida2,19. Con esta formulación a la dosis de 400/100mg dos veces al día en régimen de dosis múltiple se alcanza, aproximadamente a las 4h de la administración, una concentración máxima media en el estado de equilibrio (Cssmax) de 12,3±5,4μg/ml2.
El LPV se une a proteínas plasmáticas, principalmente a la α1-glucoproteína ácida y a la albúmina, en aproximadamente un 98-99%2, presentando a pesar de ello un elevado volumen aparente de distribución, con valores entre 70 y 130l15,20,21, que pone de manifiesto su elevado grado de acceso a órganos y tejidos. Así, esta fracción de LPV no unido a proteínas plasmáticas (1-2%) es capaz de atravesar la barrera hematoencefálica, y la concentración mínima media en el estado de equilibrio (Cssmin) de LPV en el fluido cerebroespinal es superior al cociente inhibitorio 50% para el VIH, con una relación concentración líquido cefalorraquídeo/plasma de 0,2322. Sin embargo, su acceso al aparato reproductor masculino y femenino, así como a la leche materna, es limitado23–26 y apenas atraviesa la barrera placentaria, por lo que la exposición del feto al fármaco es mínima27.
El mecanismo fundamental de eliminación de LPV es la biotransformación a nivel hepático y, en menor grado, intestinal, lo que podría condicionar su baja biodisponibilidad por un marcado efecto de primer paso. Así, el LPV experimenta una rápida e intensa oxidación vía citocromo P450 (CYP), principalmente por el CYP3A (CYP3A4 y CYP3A5), sin que existan evidencias de que presente reacciones de conjugación en fase II2,28. Se han identificado unos 13 metabolitos que se eliminan por la orina y las heces2,28.
El LPV se asocia con el RTV, conocido inhibidor enzimático de las isoenzimas CYP3A4 y CYP3A5, con el objetivo de reducir su metabolismo y mantener concentraciones terapéuticas con menores dosis, efecto conocido como «potenciación»29,30. No obstante, el RTV también es capaz de inducir su propio metabolismo, así como otras enzimas metabólicas minoritarias responsables de la biotransformación de LPV29,30. Aproximadamente un 20% y menos del 3% de la dosis de LPV en forma inalterada se excreta por vía fecal y renal, respectivamente2.
Cuando el LPV/r se administra en régimen de dosis múltiples (400/100mg) dos veces al día (BID) se alcanzan Cssmin de LPV de 8,1±5,7 μg/ml2. Su grado medio de exposición evaluado mediante el ABC en un intervalo de 12h resultó ser de 113,2±60,5 μg·h/ml2. En esta situación de equilibrio se han estimado valores medios para la semivida de eliminación y el aclaramiento oral del LPV de 5-6h y 6-7l/h, respectivamente2.
El perfil PK del LPV, coadministrado con RTV, ha demostrado ser similar entre voluntarios sanos y pacientes infectados por el VIH, así como entre pacientes naïve y pretratados a dosis estándar de 400/100mg (LPV/r) BID vía oral2,31,32. También se ha demostrado que la exposición sistémica al LPV en el estado de equilibrio es similar con la dosis de 800/200mg administrada en una dosis única (QD) o BID en pacientes naïve2,32. Además, no se han apreciado diferencias importantes en la exposición al fármaco ligadas a las diferentes formulaciones: cápsulas blandas, solución oral (ambas en presencia de alimentos) y comprimidos2,19,21,33.
Situación actual del LPV/r en la terapéuticaEl LPV sigue siendo el único IP coformulado con bajas dosis de RTV (IP/r) disponible. Además, la formulación pediátrica y la solución oral facilitan su utilización en niños y en pacientes con dificultades de deglución y permiten la individualización de las dosis.
En las guías americanas más recientes, el LPV/r ha sido desplazado por el darunavir/r y el atazanavir/r como tercer componente en el inicio del TAR en el paciente naïve6,7. La asociación británica de VIH (BHIVA) tampoco lo recomienda en esta situación, propone el efavirenz como tercer componente y reserva los IP para los casos de resistencias a inhibidores de la transcriptasa inversa análogos de nucleósido (ITIAN) y/o no análogos (ITINN), pacientes con patología neuropsiquiátrica o mujeres gestantes. Sin embargo, en el último Documento de consenso de Gesida y PNS sobre el TAR del adulto, el LPV/r se sigue recomendando como IP de primera elección en pacientes naïve, al igual que el darunavir/r y el atazanavir/r.
La experiencia en el uso del LPV/r en terapia de rescate se ha obtenido de los ensayos clínicos realizados en el resto de IP/r, que utilizan el LPV/r como IP de referencia. Tanto el tipranavir/r como el darunavir/r han demostrado superioridad respecto al LPV/r en el rescate5–7.
En la actualidad, el LPV/r combinado con otros ART continúa siendo una alternativa coste-efectiva tanto en algunos pacientes naïve como en pretratados. Además, ha demostrado un uso seguro en poblaciones especiales, como niños y embarazadas. A pesar de la disponibilidad de nuevos fármacos, la amplia experiencia clínica obtenida hasta la fecha con LPV/r es de gran valor en terapéutica y garantiza, al menos en un futuro próximo, su continuidad como uno de los IP de elección en determinadas circunstancias.
Estrategias para optimizar la terapiaMonitorización de niveles de fármacosHasta la fecha, aunque la TDM ha demostrado ser útil en la optimización de la terapia ART en determinados pacientes, existe cierta controversia sobre su uso generalizado en la práctica clínica diaria5–9,34. De hecho, existen guías de consenso en las que se recomienda la TDM en situaciones concretas en las que se prevé una alteración en la PK del LPV/r5–9.
La base fundamental que justifica la TDM es la existencia de una correlación aceptable entre concentraciones plasmáticas y respuesta. No obstante, en ocasiones esta correlación no es fácil de establecer, en especial cuando se trata de terapias combinadas como en el caso del TAR, donde el IP/r normalmente se asocia con dos ITIAN. Este hecho condiciona que los resultados de correlación concentración-respuesta terapéutica varíen según el esquema de TAR y del tipo de paciente35–38. En cuanto a toxicidad, aunque no existe una Cmax consensuada para este fármaco, sí parece demostrada una relación entre niveles plasmáticos elevados de LPV y algunos efectos secundarios39–41, aunque también se verá influida por los factores antes mencionados y la presencia de otros fármacos no ART asociados al tratamiento.
A continuación se describen las situaciones en las que actualmente está justificada la TDM de LPV/r.
Durante el inicio y control del tratamientoHabitualmente se inicia el tratamiento con LPV/r a dosis estándar de 400/100mg BID2, la cual puede no ser adecuada para los pacientes cuyo comportamiento PK se aleje de la media de la población. La determinación de las concentraciones alcanzadas en el paciente al menos una semana después de iniciado el tratamiento, para garantizar el estado de equilibrio, permitirá conocer si son superiores a los límites recomendados de 1 o 4-5,7μg/ml en pacientes naïve y pretratados, respectivamente35,40,42–46. El resultado obtenido permitirá la detección precoz de concentraciones inadecuadas que pueden dar lugar a la ineficacia del tratamiento, al desarrollo de resistencias o a la presencia de efectos adversos que suelen conducir a una disminución de la satisfacción del paciente con el tratamiento y, en consecuencia, a un mayor riesgo de discontinuación de la terapia47. Además, la estimación individualizada de los parámetros PK del LPV/r permitirá establecer, junto con la evolución clínica, la dosis más adecuada para alcanzar una respuesta óptima en el paciente. Controles sistemáticos periódicos de la respuesta clínica y de las concentraciones plasmáticas de LPV/r serán necesarios para asegurar la correcta instauración de la dosis de este fármaco45,48,49. De este modo, la TDM ayudaría a verificar si el tratamiento con LPV/r es o no adecuado para el paciente, evitando cambiar precozmente a otras combinaciones de fármacos y así preservar intactas futuras posibilidades de tratamiento.
En el control de la adherencia al tratamientoEstá ampliamente documentado que la eficacia del TAR está condicionada por la correcta adherencia al tratamiento50, la cual es difícil de conseguir en un tratamiento crónico complejo que a menudo provoca importantes efectos adversos51. Concentraciones plasmáticas de LPV anormalmente bajas y —cuando se dispone de varias concentraciones por paciente— un coeficiente de variación del índice nivel/dosis superior al 100% podrían alertar de la existencia de un problema de adherencia52. Aunque la TDM se considera una medida directa de la adherencia, no está exenta de problemas en su detección ya que, debido a la corta semivida de eliminación del LPV/r, un cumplimiento adecuado en los días previos al control de la TDM falsearía los resultados de la adherencia, al observarse las concentraciones de equilibrio que realmente alcanzaría el paciente adherente, fenómeno conocido como «adherencia de bata blanca»53,54. Por ello, los resultados de la TDM no deben interpretarse aisladamente para evaluar la adherencia, sino junto con otras medidas indirectas, tales como los registros de dispensación o de apertura del envase, cuestionarios SMAQ o entrevistas con el paciente54,55.
La TDM, al individualizar las dosis, evitaría concentraciones tóxicas y contribuiría a mejorar el grado de satisfacción del paciente con el tratamiento e indirectamente también la adherencia al mismo56.
En la identificación de interacciones con otros fármacosEl paciente infectado por el VIH frecuentemente está tratado con otros fármacos utilizados para la prevención o tratamiento de infecciones oportunistas, tratamiento sintomático y/o preventivo de los efectos adversos de los ART, etc., algunos de los cuales son sustratos, inductores o inhibidores de las enzimas responsables de las mismas vías metabólicas que el LPV/r. Así, cuando se asocia al TAR un nuevo fármaco con potencial riesgo de interacción, la TDM proporciona información sobre los niveles de LPV/r antes y después de la asociación, es decir, identifica el sentido e intensidad de la interacción y, de acuerdo con ella, permite establecer la dosis más adecuada para obtener concentraciones terapéuticas de este fármaco45,49,53,57,58. Por otra parte, el LPV/r puede también inhibir el metabolismo de otros fármacos en cuya eliminación esté implicado el CYP3A. En algunos casos este efecto da lugar a un aumento en las concentraciones plasmáticas de otros fármacos asociados que causan efectos graves que incluso pueden amenazar la vida del paciente, contraindicándose su administración conjunta. Esta situación se presenta en fármacos tales como el astemizol, la terfenadina, el midazolam, el triazolam, la cisaprida, la pimozida, la amiodarona, los alcaloides ergotamínicos, la lovastatina, la simvastatina, el vardenafilo y otros medicamentos a base de plantas que contengan hierba de San Juan (Hypericum perforatum)2.
En la detección y prevención de la toxicidadAunque en general el tratamiento con LPV/r es bien tolerado, con frecuencia aparecen problemas gastrointestinales (diarreas, náuseas y vómitos) y algunas complicaciones metabólicas como la dislipemia, resistencia a la insulina y lipodistrofia39–41,59. También se ha descrito una prolongación del intervalo PR y QT, y el aumento del riesgo de hemorragias en pacientes hemofílicos2. Considerando que algunos estudios han encontrado una cierta relación entre niveles elevados de LPV/r y presencia de efectos adversos39–41,49,60, la TDM permitiría detectar situaciones de sobredosificación que podrían resolverse reduciendo adecuadamente la dosis de este medicamento y así evitar un cambio de tratamiento por intolerancia53. No obstante, algunos investigadores también sugieren la utilidad de la TDM en pacientes sin manifestaciones aparentes de toxicidad pero que presentan niveles elevados45. Ello es debido a que algunos efectos adversos provocados por el TAR, como las alteraciones metabólicas, suelen aparecer de forma más tardía y en consecuencia podrían ser prevenibles mediante la TDM. En cualquier caso, independientemente de los argumentos que sugieran una reducción de dosis, es importante considerar la concentración diana óptima a alcanzar, la cual varía en pacientes naïve respecto a los pretratados, con valores de Cssmin de 1 o de 4μg/ml, respectivamente, lo que contribuye a minimizar los riesgos de ineficacia clínica y posible desarrollo de resistencias35,40,42–46.
En poblaciones especialesDurante el embarazo se pueden modificar los procesos de absorción-distribución-metabolismo-excreción (ADME)45, dando lugar a cambios significativos en las concentraciones plasmáticas de LPV/r. Existe evidencia de que la exposición sistémica a este medicamento se reduce durante el tercer trimestre de gestación27. Por ello en mujeres clínicamente controladas antes del embarazo, se recomienda obtener niveles de LPV/r previos al segundo trimestre de gestación como referencia, los cuales deberán mantenerse durante el resto del embarazo mediante los ajustes posológicos necesarios53.
La población pediátrica es un grupo muy heterogéneo en el que el comportamiento PK del LPV/r difiere de la población adulta. A pesar de las recomendaciones posológicas para niños y adolescentes teniendo en cuenta la edad, el peso y la superficie corporal, no se puede asegurar la misma exposición al fármaco que en adultos45,53. Aunque la información disponible acerca de la PK de ART en pacientes pediátricos se está incrementando paulatinamente, los datos PK y de eficacia y seguridad del LPV/r en niños menores de 2 años aún son limitados, por lo que su utilización no está recomendada2. En este sentido, la TDM cada vez más utilizada en niños y recomendada en las guías PENTA (Pediatric European Network for Treatment of AIDS) puede ayudar a conseguir un grado de exposición adecuado en este sector de la población.
En pacientes con pesos extremos también está recomendada la TDM, debido a los potenciales riesgos de toxicidad e ineficacia clínica que puede presentar esta población15. Aunque el género por sí mismo no parece afectar el comportamiento PK del LPV/r, el hecho de que con frecuencia las mujeres presenten menor peso corporal puede dar lugar a mayores niveles de estos fármacos cuando reciben la dosis estándar, recomendándose también en ellas la monitorización45.
Los pacientes con daño hepático son otros candidatos que pueden beneficiarse de la TDM del LPV/r debido al alto grado de metabolismo hepático que experimenta este medicamento28,45. En estos pacientes puede incrementase la exposición sistémica a la fracción libre de LPV/r, debido a una reducción en el metabolismo y en el grado de unión a proteínas plasmáticas61. No obstante, ya que habitualmente se determinan las concentraciones plasmáticas de fármaco total, su interpretación puede ser errónea en situaciones en las que la fracción de fármaco libre esté alterada61. Aunque hay algoritmos predictivos de las concentraciones de estos fármacos en función del daño hepático, presentan un alto grado de incertidumbre45. Además, no se ha establecido la eficacia y la seguridad del LPV/r en pacientes con trastornos hepáticos subyacentes significativos2.
Entre los pacientes infectados por el VIH existen casos de coinfectados con el virus de la hepatitis B o C, con un mayor riesgo de desarrollar fallo hepático secundario al tratamiento con LPV/r39,62. En esta situación, la dosis estándar administrada podría resultar potencialmente tóxica, por lo que se recomienda monitorizar los niveles plasmáticos de este IP47,48,56.
En pacientes con insuficiencia renal, debido a la baja contribución del riñón a la eliminación del LPV/r, no se esperan modificaciones significativas en sus concentraciones plasmáticas2.
En regímenes de dosificación fuera de indicaciónEn esquemas de tratamiento con LPV/r diferentes a los recomendados en las condiciones de autorización, como es la monoterapia, la TDM puede ser una herramienta que asegure concentraciones de LPV/r adecuadas a cada paciente17,48.
A pesar de la evidencia actual sobre el beneficio de la TDM como herramienta de ayuda en la optimización de la terapia con LPV/r, son necesarias futuras investigaciones que evalúen las prestaciones de esta estrategia. Así, sería importante disponer de mayor información sobre márgenes terapéuticos de LPV/r que tomen en consideración el tipo de TAR, así como el genotipo del virus y sus parámetros PK en poblaciones específicas de VIH para mejorar la capacidad predictiva de los algoritmos bayesianos. La identificación de polimorfismos genéticos (single nucleotide polymorphism, SNP) que afecten la PK del LPV/r, revisados en el siguiente apartado, también resultan de interés para su incorporación, como factores de disposición, en el análisis PK34.
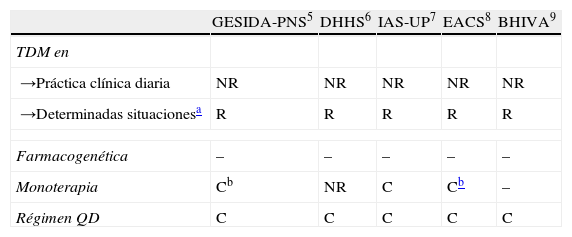
La tabla 1 recoge las recomendaciones de las guías nacionales e internacionales de consenso sobre el uso de la TDM del LPV/r.
Indicaciones en las principales guías nacionales e internacionales sobre las estrategias de optimización posológica de lopinavir/ritonavir evaluadas en el estudio
| GESIDA-PNS5 | DHHS6 | IAS-UP7 | EACS8 | BHIVA9 | |
| TDM en | |||||
| →Práctica clínica diaria | NR | NR | NR | NR | NR |
| →Determinadas situacionesa | R | R | R | R | R |
| Farmacogenética | – | – | – | – | – |
| Monoterapia | Cb | NR | C | Cb | – |
| Régimen QD | C | C | C | C | C |
GESIDA-PNS: Grupo de estudio del SIDA-SEIMC - Plan Nacional sobre el Sida; DHHS: Department of Health and Human Services; IAS-UP: International AIDS Society-USA Panel; EACS: European AIDS Clinical Society; BHIVA: British HIV Association; TDM: monitorización terapéutica de fármacos; R: recomendado; C: considerar; NR: no recomendado; –: Sin información; QD: administración una vez al día.
La administración de una misma dosis de un fármaco ART a un grupo de pacientes generalmente da lugar a una elevada variabilidad interindividual en la eficacia y la toxicidad del TAR, que puede atribuirse en parte a sus diferencias demográficas y clínicas. Además, entre las causas responsables de esta variabilidad, las variaciones genéticas pueden constituir un factor significativo63. Así, se ha demostrado que la presencia de variaciones genéticas en genes codificantes de ciertas proteínas implicadas en el metabolismo del LPV/r (CYP3A4, CYP3A5, etc.) y en su transporte (MRP-2, SLCO) pueden influir en su comportamiento PK15,64–66. Por otra parte, la presencia de SNP en los genes que codifican proteínas implicadas en el metabolismo lipídico (APOA5, APOC3, TNF, SREBP1, etc.) afectará en mayor o menor medida al grado en el que se puede manifestar el síndrome metabólico atribuido a IP/r67–70.
Para que la información genética pueda aplicarse en clínica, los genes analizados deben ejercer un efecto dominante, la influencia del genotipo sobre los efectos del tratamiento prescrito debe ser significativa y el coste-efectividad de su aplicación debe estar demostrado9. Para el caso del LPV/r aún es preciso establecer claramente la relación entre estos SNP y su comportamiento PK, ya que la mayoría de los estudios se han centrado en el efecto de SNP de un solo gen, mientras que en estos procesos de ADME están involucrados múltiples genes relacionados entre ellos junto con otros factores no genéticos.
Debido al gran interés que está despertando la PG y al número creciente de casos clínicos publicados en los que se demuestra su utilidad en la práctica clínica10,71, se están realizando diversas investigaciones con los diferentes ART para buscar variaciones genéticas que muestren relación significativa con su eficacia y/o toxicidad. Sin embargo, en el caso de los IP, existen actualmente pocos estudios que evidencien de manera consistente relaciones de este tipo.
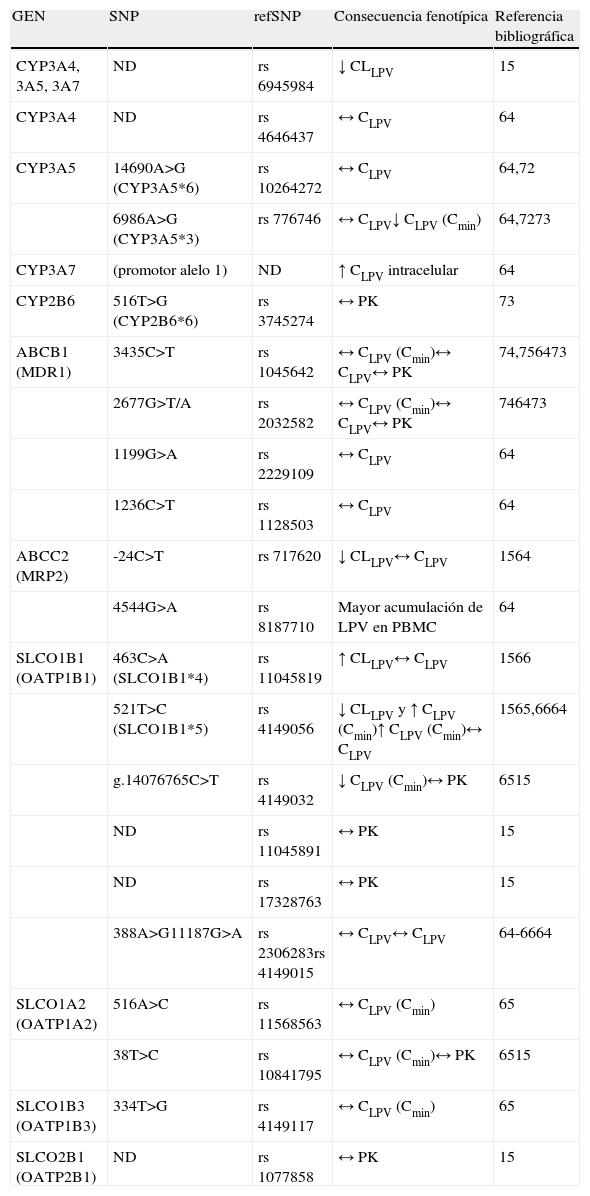
En la tabla 2 se muestran los resultados de las variaciones genéticas con posible influencia sobre la PK del LPV/r de los estudios realizados hasta la fecha, indicando el rs cuando está disponible y el SNP. Aunque existen indicios, la relación entre los SNP de CYP3A4, CYP3A5 y SLCO y la PK del LPV/r todavía no está suficientemente documentada, por lo que se precisan más estudios de investigación clínica que la confirmen. Estos hallazgos resultarían de gran utilidad para la prescripción a priori de las dosis más adecuadas a las características genéticas del paciente y se integrarían con la información PK en los modelos de población15,72-75.
Influencia de polimorfismos genéticos de enzimas y transportadores sobre la farmacocinética del lopinavir/ritonavir
| GEN | SNP | refSNP | Consecuencia fenotípica | Referencia bibliográfica |
| CYP3A4, 3A5, 3A7 | ND | rs 6945984 | ↓ CLLPV | 15 |
| CYP3A4 | ND | rs 4646437 | ↔ CLPV | 64 |
| CYP3A5 | 14690A>G (CYP3A5*6) | rs 10264272 | ↔ CLPV | 64,72 |
| 6986A>G (CYP3A5*3) | rs 776746 | ↔ CLPV↓ CLPV (Cmin) | 64,7273 | |
| CYP3A7 | (promotor alelo 1) | ND | ↑ CLPV intracelular | 64 |
| CYP2B6 | 516T>G (CYP2B6*6) | rs 3745274 | ↔ PK | 73 |
| ABCB1 (MDR1) | 3435C>T | rs 1045642 | ↔ CLPV (Cmin)↔ CLPV↔ PK | 74,756473 |
| 2677G>T/A | rs 2032582 | ↔ CLPV (Cmin)↔ CLPV↔ PK | 746473 | |
| 1199G>A | rs 2229109 | ↔ CLPV | 64 | |
| 1236C>T | rs 1128503 | ↔ CLPV | 64 | |
| ABCC2 (MRP2) | -24C>T | rs 717620 | ↓ CLLPV↔ CLPV | 1564 |
| 4544G>A | rs 8187710 | Mayor acumulación de LPV en PBMC | 64 | |
| SLCO1B1 (OATP1B1) | 463C>A (SLCO1B1*4) | rs 11045819 | ↑ CLLPV↔ CLPV | 1566 |
| 521T>C (SLCO1B1*5) | rs 4149056 | ↓ CLLPV y ↑ CLPV (Cmin)↑ CLPV (Cmin)↔ CLPV | 1565,6664 | |
| g.14076765C>T | rs 4149032 | ↓ CLPV (Cmin)↔ PK | 6515 | |
| ND | rs 11045891 | ↔ PK | 15 | |
| ND | rs 17328763 | ↔ PK | 15 | |
| 388A>G11187G>A | rs 2306283rs 4149015 | ↔ CLPV↔ CLPV | 64-6664 | |
| SLCO1A2 (OATP1A2) | 516A>C | rs 11568563 | ↔ CLPV (Cmin) | 65 |
| 38T>C | rs 10841795 | ↔ CLPV (Cmin)↔ PK | 6515 | |
| SLCO1B3 (OATP1B3) | 334T>G | rs 4149117 | ↔ CLPV (Cmin) | 65 |
| SLCO2B1 (OATP2B1) | ND | rs 1077858 | ↔ PK | 15 |
SNP: single nucleotide polymorphism; LPV: lopinavir; PBMC: células de sangre periférica mononucleadas; ND: no disponible; ↓: disminución de; ↑: incremento de; ↔: sin influencia en; CLLPV: aclaramiento de lopinavir; PK: farmacocinética; CLPV: concentración de lopinavir; Cmin: concentración mínima.
Actualmente se intenta conseguir tratamientos con la misma eficacia obtenida hasta ahora pero con menor toxicidad y menor coste. Los IP/r presentan un favorable perfil PK, una alta barrera genética al desarrollo de resistencias y un elevado cociente inhibitorio, lo que los convierte en excelentes candidatos para su uso en monoterapia76–79. Esta estrategia permite a su vez mantener intactas otras opciones terapéuticas16 y puede estar asociada con una menor toxicidad a largo plazo, como la lipodistrofia, al retirarse los otros componentes del TAR80.
La simplificación del tratamiento con dos ITIAN más LPV/r a la monoterapia con este último ha mostrado seguridad y eficacia en un alto porcentaje de pacientes en varios estudios realizados en la última década81–85. En caso de fracaso virológico, asociado con frecuencia a problemas puntuales de adherencia y excepcionalmente al desarrollo de resistencias al LPV/r83, la estrategia de rescate normalmente cursa con el restablecimiento de los ITIAN que el paciente tomaba previamente81–84.
Con el IP darunavir/r también se han llevado a cabo estudios aleatorizados, tanto en régimen BID como QD86,87, y por primera vez se ha demostrado la no inferioridad de monoterapia con un IP/r en régimen QD (darunavir/r) frente a su uso con dos ITIAN86. Los datos con atazanavir/r son insuficientes, al igual que con LPV/r en régimen QD88. Cabe destacar que España ha sido pionera, junto con otros países, en el estudio de esta nueva estrategia de simplificación del TAR a monoterapia con LPV/r, donde se han llevado a cabo algunas de las primeras y más importantes investigaciones hasta el momento81,83,89.
A pesar de que no existe consenso en las recomendaciones de la monoterapia con IP/r en las diferentes guías internacionales sobre el TAR, está demostrado que una gran proporción de pacientes pueden mantener la supresión viral con LPV/r o darunavir/r en monoterapia. La sociedad europea del sida8 considera la posibilidad de la monoterapia con LPV/r o darunavir/r como estrategia de simplificación en pacientes en los que exista intolerancia a los ITIAN, que no presenten resistencias a IP y que hayan tenido la carga viral suprimida al menos en los 6 meses anteriores. La sociedad internacional del sida7 la recomienda en situaciones muy concretas en las que otros fármacos ART no pueden utilizarse debido a problemas de toxicidad; sin embargo, el departamento de salud y servicios humanos estadounidense (DHHS) no recomienda la monoterapia en ninguna situación6. En la actualidad se están desarrollando otros importantes estudios sobre monoterapia de IP/r frente a triterapia, y sus resultados podrían ayudar a los expertos a definir la utilización óptima de esta estrategia90,91.
Administración en régimen QDEn la práctica clínica diaria hay constancia de la administración de algunos IP en régimen QD en pacientes naïve que iniciaban TAR para facilitar la adherencia, aunque la recomendación autorizada fuera el régimen BID9. En el caso del LPV/r, la dosificación QD (800/200mg) ya ha sido autorizada por la EMA y la FDA. Aunque la dosis total diaria de LPV/r es la misma para los dos regímenes, se producen cambios sustanciales en la evolución de las concentraciones de LPV/r, incrementándose significativamente las fluctuaciones entre las Cssmax y Cssmin. Esta situación puede ser clínicamente relevante, ya que incrementos en las Cssmax se relacionan directamente con alteraciones gastrointestinales17,92 y descensos de las Cssmin por debajo de la concentración mínima eficaz pueden comprometer la eficacia del TAR93, favoreciendo la aparición de resistencias al fármaco17,92. No obstante, estudios recientes han mostrado similar eficacia, perfil de tolerancia y desarrollo de resistencias en ambos regímenes de dosificación, tanto en pacientes naïve para el TAR como pretratados94,95. Como era de esperar, se ha observado un incremento en la adherencia de los pacientes que toman el régimen QD frente al BID93–95.
ConclusionesAlgunas guías de consenso siguen recomendando el LPV/r como tercer componente del TAR en el paciente naïve; sin embargo, en pacientes con cierto grado de resistencias a IP no está indicado como fármaco de elección. Para optimizar la utilización del LPV/r en terapéutica, existen diferentes estrategias en la práctica clínica, como la TDM. Esta podría ser una herramienta útil para realizar ajustes posológicos de LPV/r en determinadas situaciones donde se modifica su PK, para garantizar concentraciones adecuadas del fármaco. Así mismo, el análisis de la influencia de SNP de enzimas metabolizadoras y proteínas transportadoras de LPV/r sobre su cinética de disposición, podría ayudar a esclarecer la alta variabilidad interindividual de las concentraciones observadas cuando se administra el fármaco a dosis estándar. Finalmente, el nuevo esquema posológico que utiliza este fármaco en monoterapia está demostrando una favorable relación riesgo-beneficio, aunque no existe consenso entre las guías internacionales sobre esta indicación para LPV/r.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

