




Las tetraciclinas, naturales o semisintéticas, actúan inhibiendo la síntesis de las proteínas bacterianas. Son bacteriostáticas, con amplio espectro de actividad. Estigmatizadas tiempo atrás por la frecuencia de microorganismos resistentes, actualmente han renacido al recuperar sensibilidad e incorporarse nuevos y más activos componentes. La doxiciclina es la tetraciclina más utilizada actualmente y constituye uno de los medicamentos esenciales de la Organización Mundial de la Salud. La tigeciclina, una tetraciclina de tercera generación, tiene un mayor espectro de actividad, y representa una alternativa en el tratamiento de infecciones complicadas con microorganismos multirresistentes. Las sulfamidas son antibióticos sintéticos, bacteriostáticos, de amplio espectro. Debido a su toxicidad y elevada resistencia su uso actualmente es muy escaso. El metronidazol es el principal componente de la familia de los 5-nitroimidazoles. Es un antibiótico con gran actividad bactericida frente a anaerobios y algunos microaerófilos y continúa siendo muy útil en el tratamiento de infecciones bacterianas y parasitarias.
Tetracyclines, whether natural or semisynthetic, act by inhibiting bacterial protein synthesis. These antibiotics are bacteriostatic and have a broad spectrum of activity. In the past, tetracyclines were discredited because of the high prevalence of isolates with acquired resistance, but they are now regaining status because of the incorporation of new, more active components. Doxycycline is currently the most commonly used tetracycline and is considered an essential drug by the World Health Organization. Tigecycline, a third-generation tetracycline, has a broader spectrum of activity and is an alternative option for the treatment of complicated infections by multiresistant organisms. Sulfonamides are synthetic, broad-spectrum bacteriostatic antibiotics. Because of associated toxicity and high rates of resistance, their use is now very limited. Metronidazole is the most important member of the 5-nitroimidazole family. It has high bactericidal activity against anaerobic bacteria, some microaerophilic bacteria and protozoa, and remains very useful in the treatment of bacterial and parasitic infections.
Las tetraciclinas constituyen una familia de productos naturales (clortetraciclina, oxitetraciclina, tetraciclina, demeclociclina) y semisintéticos (metaciclina, doxiciclina, minociclina, limeciclina, rolitetraciclina, tigeciclina, PTK 7906) derivados de diferentes especies de Streptomyces spp. Actúan inhibiendo la síntesis de las proteínas bacterianas mediante la unión a la subunidad ribosomal 30S de las bacterias. Son agentes básicamente bacteriostáticos, con actividad frente a una gran variedad de microorganismos, por lo que se convirtieron en antibióticos de uso habitual tanto en seres humanos como en animales, y también se utilizaron en algunas áreas de la agricultura1–5. El espectro antimicrobiano relativamente limitado de las tetraciclinas clásicas, la imposibilidad de utilizarse en niños, durante el embarazo y la lactancia, y la aparición de nuevos componentes más eficaces en otras familias de antibióticos, han ocasionado que el uso de tetraciclinas en humanos, con algunas excepciones, sea escaso. En España, el consumo de tetraciclinas ha ido descendiendo paulatinamente con los años, de forma que el número de dosis diarias definidas por 1.000 habitantes y día descendió desde 0,8 en el año 1998 a 0,6 en el año 20066. Este consumo se debió casi exclusivamente al uso de doxiciclina, la tetraciclina más utilizada en humanos en todo el mundo, y uno de los medicamentos esenciales de la Organización Mundial de la Salud7. El desarrollo de nuevas tetraciclinas semisintéticas, cuyo principal exponente es la tigeciclina, puede hacer variar este panorama en los próximos años. La Food and Drug Administration americana aprobó en el año 2005 la comercialización de tigeciclina, primera tetraciclina desarrollada dentro de la nueva clase de las glicilciclinas, con estructura de tetraciclina derivada de la minociclina. Un año después, la Agencia Española del Medicamento aprobó su comercialización, y se encuentra indicada en el tratamiento de infecciones complicadas de piel y tejidos blandos y en infecciones intraabdominales complicadas. En España, además de doxiciclina y tigeciclina, se comercializan para uso humano oxitetraciclina, tetraciclina y minociclina. Los otros componentes naturales y semisintéticos se han retirado o no se han llegado a comercializar.
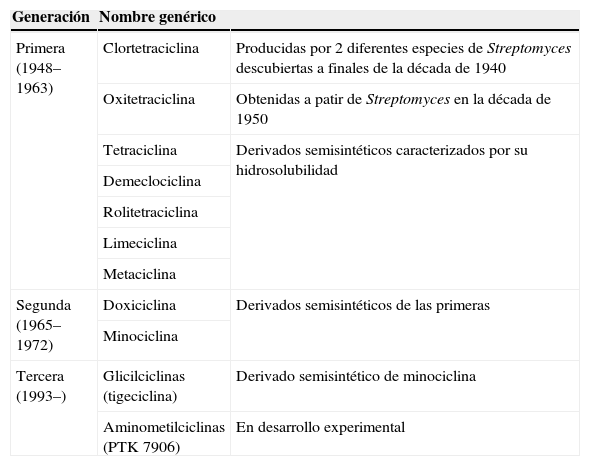
Clasificación y estructura químicaDe acuerdo con el orden de descubrimiento, las propiedades farmacocinéticas y el espectro de actividad antimicrobiana las tetraciclinas pueden dividirse en 3 grupos o generaciones (tabla 1).
Principales componentes del grupo de las tetraciclinas
| Generación | Nombre genérico | |
| Primera (1948–1963) | Clortetraciclina | Producidas por 2 diferentes especies de Streptomyces descubiertas a finales de la década de 1940 |
| Oxitetraciclina | Obtenidas a patir de Streptomyces en la década de 1950 | |
| Tetraciclina | Derivados semisintéticos caracterizados por su hidrosolubilidad | |
| Demeclociclina | ||
| Rolitetraciclina | ||
| Limeciclina | ||
| Metaciclina | ||
| Segunda (1965–1972) | Doxiciclina | Derivados semisintéticos de las primeras |
| Minociclina | ||
| Tercera (1993–) | Glicilciclinas (tigeciclina) | Derivado semisintético de minociclina |
| Aminometilciclinas (PTK 7906) | En desarrollo experimental |
PTK 7906: 7-dimetilamino, 9-aminometilciclina.
Primera generación: la constituyen los agentes más antiguos. Son los menos lipofílicos y los que peor absorción muestran. Aquí se incluyen tetraciclina, oxitetraciclina, clortetraciclina, demeclociclina, limeciclina, metaciclina y rolitetraciclina. Todos ellos, excepto rolitetraciclina, pueden administrarse por vía oral.
Segunda generación: presentan una mejor absorción y son entre 3 y 5 veces más lipofílicos que los componentes del grupo anterior. En este grupo se incluyen doxiciclina y minociclina. Se pueden administrar por vía oral y también por vía intravenosa.
Tercera generación: las glicilciclinas pertenecen a la última generación de tetraciclinas. Son análogos semisintéticos obtenidos tras modificar la posición 9 del anillo tetracíclico de los compuestos de las generaciones anteriores. La tigeciclina es el 9-tert-butil-glicilamido derivado de la minociclina y constituye el principal representante de este nuevo grupo. Además de las glicilciclinas, en esta tercera generación se incluyen nuevos compuestos en desarrollo, como las aminometilciclinas, de cuyo grupo ya ha pasado a experimentación humana la PTK 0796 para administración oral e intravenosa en una sola dosis diaria.
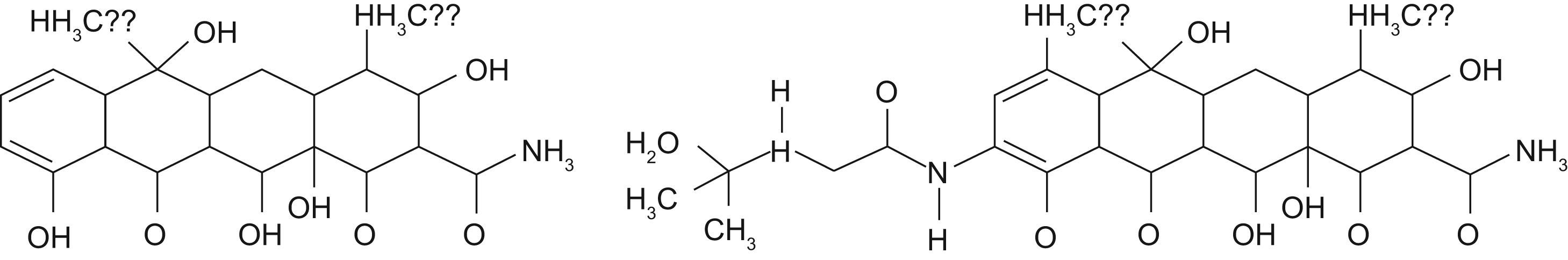
Todas las tetraciclinas poseen un núcleo de estructura tetracíclica lineal compuesta de 4 anillos fusionados (fig. 1). Todos forman complejos quelantes con distintos cationes, como calcio, magnesio o hierro, lo que los hace insolubles en agua y dificultan su absorción.
Farmacocinética
Primera y segunda generación: principalmente se administran por vía oral, aunque existen algunos compuestos que también pueden administrarse por vía intravenosa (tetraciclina, oxitetraciclina, doxiciclina y minociclina). Rolitetraciclina se administra exclusivamente por vía intravenosa. No es habitual la vía intramuscular debido al intenso dolor que produce su inyección. Los que se administran por vía oral se absorben de manera variable en el estómago y en el intestino delgado, dependiendo de la tetraciclina considerada. Las de primera generación se absorben peor, con un rango de absorción que oscila entre el 25–60%. La doxiciclina y la minociclina son las que mejor se absorben (90–100%) pues en ellas no interfiere de forma significativa la alimentación. El resto se absorben peor (≤80%), por lo que deben administrarse fuera de las comidas. El hierro y otros medicamentos o compuestos que formen complejos quelantes o aumenten el pH del estómago (calcio, aluminio, bismuto, magnesio, cimetidina y omeprazol) interfieren en su absorción.
Las concentraciones máximas séricas dependen de la dosis administrada. Tras una dosis oral normal son de 1,5–6,0μg/ml. El volumen de distribución de estos agentes oscila entre 0,7l/kg para la doxiciclina hasta 1,7l/kg para la demeclociclina. La unión con las proteínas es variable: la doxiciclina y la minociclina (un 60–95 y un 55–76%, respectivamente) tienen mayor unión a las proteínas que la tetraciclina (20–65%). Difunden ampliamente en todos los tejidos y líquidos por su gran liposolubilidad, en particular las de acción larga. En el líquido cefalorraquídeo (LCR) las tetraciclinas alcanzan niveles del 10–26% de los séricos. En el esputo, las concentraciones oscilan entre el 8–20% y son suficientes para inhibir neumococos y Haemophilus influenzae sensibles. Penetran en el sebo y se eliminan a través del sudor, por lo que están indicadas en el tratamiento del acné. La penetración en saliva es baja y las concentraciones en el hígado, el riñón y el aparato digestivo son altas. Se acumulan en los huesos y los dientes, pasan la barrera fetoplacentaria y se excretan, habitualmente en elevadas concentraciones, en la leche materna. Según su perfil farmacocinético pueden agruparse en 3 categorías: semivida corta (5–9h), clortetraciclina, oxitetraciclina y tetraciclina; semivida intermedia (10–14h), demeclociclina, limeciclina y metaciclina; semivida larga (16–18h); la doxiciclina y la minociclina son las más liposolubles. Todos los compuestos, excepto tetraciclina, que presenta un metabolismo hepático, se eliminan sin metabolizar a través de las vías biliar y renal. La concentración en la bilis es entre 5 y 25 veces superior a la concentración sérica. La eliminación por orina varía según el compuesto, y es muy escasa para minociclina (6%) y clortetraciclina (18%), moderada-baja para doxiciclina (42%) y aceptable para tetraciclina (60%), por lo que con la posible excepción de las 2 primeras se alcanzan concentraciones terapéuticas en la orina para el tratamiento de infecciones urinarias por microorganismos sensibles. El resto del fármaco se elimina en las heces. La doxiciclina es la tetraciclina habitualmente recomendada en pacientes con infección extrarrenal y fallo renal.
Tercera generación: tigeciclina. Solo puede administrarse por vía intravenosa en infusión durante 30–60min. El volumen de distribución es amplio, tras la dosis habitual oscila entre 2,5–7l/kg. Esto indica una buena penetración en los tejidos, que alcanza altas concentraciones en algunos órganos. Tras la administración de la pauta habitual de 100mg seguida de dosis de 50mg cada 12h, la concentración sérica alcanzada es de 0,6mg/l. La concentración pulmonar 4h después es de 0,8mg/l. La penetración en el LCR con meninges no inflamadas es del 5% tras 1 h desde la administración, y superior al 40% a las 24h. La concentración biliar es muy elevada, entre 600 y 2.000 veces la concentración sérica. El fármaco se acumula especialmente en el hueso, probablemente por su unión al calcio. También se acumula en la médula ósea, la glándula tiroides, el bazo y el hígado. La vida media es larga, y oscila entre 30 y 40h tras la dosificación habitual8.
La unión a proteínas es del 70%. Aproximadamente el 20% de la tigeciclina se metaboliza en el hígado mediante glucuronización antes de la excreción. Como sucede con otras tetraciclinas, hay una excreción biliar y una circulación enterohepática. La tigeciclina se elimina principalmente a través de las heces sin metabolizar. El resto del fármaco, alrededor del 30%, se elimina en la orina (el 15% en forma activa). Este perfil farmacocinético no se ve afectado en caso de insuficiencia renal, ni como consecuencia de la hemodiálisis, por lo que no se requiere ajuste de la dosis en estas situaciones. Tampoco es necesario el ajuste en pacientes con insuficiencia hepática leve. En pacientes con insuficiencia hepática grave, la dosis debe reducirse a 25mg cada 12h, tras la dosis inicial de 100mg.
FarmacodinamiaLa farmacodinamia de las tetraciclinas clásicas no se ha estudiado tan a fondo como la de otros antimicrobianos, pero en general, al igual que sucede con betalactámicos y macrólidos, la actividad de las tetraciclinas sigue un patrón farmacodinámico dependiente del tiempo, en el que la eficacia depende del tiempo en que la concentración tisular del antibiótico es superior a la concentración mínima inhibitoria (CMI). En el caso de la doxiciclina, esto es válido para concentraciones bajas, ya que a altas concentraciones su actividad es dependiente de la concentración. Respecto a la tigeciclina, los estudios realizados indican que para conseguir una actividad óptima su concentración debe estar por encima de la CMI al menos durante el 50% del tiempo del intervalo entre dosis.
Como sucede con otros antimicrobianos que actúan inhibiendo la síntesis de proteínas, las tetraciclinas tienen un efecto postantibiótico prolongado, que es especialmente largo en el caso de la tigeciclina. Esta característica, junto con la vida media larga, indica que el parámetro farmacodinámico más adecuado para predecir la eficacia clínica y microbiológica de la tigeciclina es el cociente entre el área bajo la curva y la CMI8.
Espectro de actividadLa eficacia de las tetraciclinas fue disminuyendo en las décadas precedentes debido a la amplia existencia de genes de resistencia, probablemente como consecuencia del prolongado y extenso uso de estos antimicrobianos en los seres humanos y como promotores del crecimiento en animales. Afortunadamente, en la década actual, la resistencia ha disminuido, y pueden recuperarse algunas de las indicaciones previamente abandonadas. Las tetraciclinas tienen en general un comportamiento antimicrobiano similar; las de tercera generación son una excepción.
Las tetraciclinas de primera y segunda generación son inicialmente activas frente a un gran espectro de bacterias grampositivas y gramnegativas, aunque el porcentaje de cepas resistentes en cada especie o género bacteriano es muy variable. Actualmente hay pocas cepas de Streptococcus pyogenes y Staphylococcus aureus resistentes a tetraciclinas en nuestro medio (<5%), y la resistencia es moderada en Staphylococcus saprophyticus (10–15%). La resistencia es tan elevada en Streptococcus agalactiae (>80%) y Enterococcus spp. que las hace inadecuadas para el tratamiento. La resistencia en Streptococcus pneumoniae ha disminuido en los últimos años (el 60–70% de cepas eran resistentes al principio de la década de 1980, mientras que en la actualidad tan sólo el 10% de los causantes de neumonía o el 20% de los aislados en las exacerbaciones de EPOC son resistentes). Este último hecho, junto con el bajísimo porcentaje de H. influenzae y Moraxella catarrhalis resistentes, permite que las tetraciclinas puedan utilizarse en alguna de las exacerbaciones agudas de los bronquíticos crónicos. Dentro de los bacilos grampositivos son sensibles Corynebacterium spp., Listeria monocytogenes, Clostridium spp. (la mayoría) y Bacillus anthracis. Nocardia spp. es poco activa;la minociclina es la más activa frente a este microorganismo. También son activas frente a los gramnegativos, aunque muestran diferentes grados de resistencia adquirida, Escherichia coli, Vibrio spp., Brucella spp., Yersinia spp., Legionella pneumophila, Plesiomonas shigelloides, Aeromonas spp. y Neisseria gonorrhoeae. En nuestro medio, muy bajo porcentaje de cepas de Helicobacter pylori presentan resistencia (<1%). Se incluyen dentro del espectro antimicrobiano algunos anaerobios como Bacteroides grupo fragilis, aunque la mayoría (>50%) de las cepas siguen siendo resistentes actualmente. Son muy activas frente a Rickettsia spp. y Coxiella burnetii, y tanto como lo puedan ser los macrólidos frente a Mycoplasma spp. y Chlamydia spp. Algunas espiroquetas (incluidas Treponema pallidum y Borrelia burgdorferi), micobacterias no tuberculosas (Mycobacterium marinum) y algunos protozoos (Plasmodium falciparum, Entamoeba histolytica y Balantidium coli) presentan sensibilidad a tetraciclinas.
La tigeciclina muestra un espectro de actividad antimicrobiana mucho más amplio que las tetraciclinas clásicas. Su espectro abarca a los microorganismos frente a los que son activas las tetraciclinas clásicas y la gran mayoría de los que han desarrollado resistencia a estos agentes, incluyendo cocos grampositivos y enterobacterias multirresistentes9,10. La tigeciclina es activa frente a S. aureus y estafilococos coagulasa negativo resistentes a meticilina y frente a cerca del 80% de las cepas de Enterococcus faecalis y Enterococcus faecium sensibles y resistentes a vancomicina. La tigeciclina presenta una buena actividad frente a bacteroides del grupo fragilis. La CMI90 de E. coli y Klebsiella pneumoniae multirresistentes, portadoras de betalactamasas de espectro ampliado es <1mg/l y 2mg/l, respectivamente11. Aunque actualmente es excepcional, se ha descrito aparición de resistencia a tigeciclina en algunas enterobacterias durante el curso del tratamiento12,13. Frente a cepas de Acinetobacter spp., la tigeciclina puede mostrar sensibilidad frente a cepas resistentes a imipenem14, aunque las cepas resistentes no son infrecuentes y también se han descrito la aparición de resistencia durante el tratamiento12,15. Existen discrepancias entre los resultados de las pruebas de susceptibilidad frente a Acinetobacter, que usan elución o dilución en agar frente a dilución en caldo; se aconseja utilizar la dilución en caldo para estudiar su susceptibilidad con cualquier tipo de tetraciclina16.
Es activa frente a Stenotrophomonas maltophilia pero menos que minociclina. Proteus, Morganella y Providencia son poco o nada sensibles. Apenas es activa frente a Pseudomonas aeruginosa y Burkholderia cepacia; no es recomendable su uso para el tratamiento de estas infecciones. Aunque existen pocos datos sobre la actividad de la tigeciclina frente a patógenos atípicos, es tanto o más activa que las tetraciclinas clásicas frente a Mycoplasma pneumoniae, Chlamydophila pneumoniae, B. burgdorferi y Nocardia spp. También es activa frente a micobacterias de crecimiento rápido, especialmente frente a las del grupo Mycobacterium chelonae-Mycobacterium abscessus17.
Mecanismo de acciónLas tetraciclinas actúan inhibiendo la síntesis proteica de las bacterias. Se fijan con gran afinidad a la subunidad 30S del ribosoma bacteriano, de manera que impiden la unión del sitio aminoacil del ácido ribonucleico de transferencia a la subunidad 30S ribosomal, y de esta forma se paraliza la incorporación de aminoácidos durante la síntesis proteica. Atraviesan la membrana externa de las bacterias a través de porinas mediante difusión pasiva y llegan al citoplasma gracias a un mecanismo dependiente de energía. Las glicilciclinas, entre las que se encuentra la tigeciclina, se fijan al mismo punto de unión del ribosoma bacteriano, pero la última lo hace con una fuerza 5 veces superior a la de la tetraciclina y la minociclina. Probablemente esta fuerza de anclaje sea la que permite a la tigeciclina vencer el mecanismo de resistencia ribosomal frente a las tetraciclinas clásicas. La fijación de todas las tetraciclinas a la subunidad ribosomal es reversible, lo que explicaría su efecto bacteriostático.
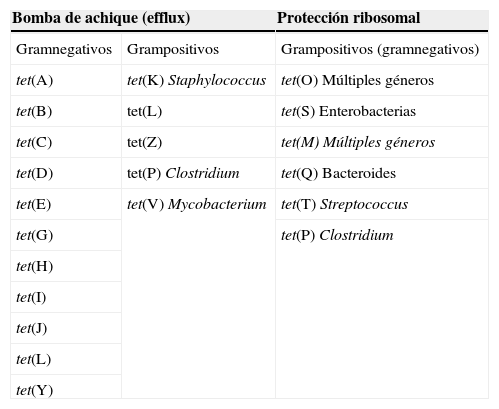
La resistencia se produce principalmente impidiendo la unión del antibiótico a la diana (protección ribosomal) o mediante la expulsión del antibiótico al exterior de la célula por medio de bombas (achique o efflux). Los determinantes de resistencia se encuentran en genes que se localizan normalmente en elementos móviles, como plásmidos, transposones conjugativos e integrones. Estos elementos móviles portadores de los genes de resistencia pueden transferirse a bacterias de la misma especie o de especies diferentes mediante conjugación y transformación bacteriana. En los integrones, junto al gen que confiere la resistencia a tetraciclinas con frecuencia se encuentran otros genes (gene cassettes) que confieren resistencia a otros antibióticos, por lo que las tetraciclinas u otros antimicrobianos pueden seleccionar estas cepas multirresistentes. Los determinantes genéticos implicados en la resistencia a tetraciclinas son los genes tet y otr. Existen genes tet que codifican resistencia tanto por bomba de achique como por modificación ribosomal (tabla 2). La disminución en la acumulación intracelular de tetraciclinas por bombeo activo asociado a la membrana (achique o efflux) es el mecanismo habitual que les confiere resistencia de forma natural o adquirida en bacterias gramnegativas. La resistencia a las tetraciclinas clásicas principalmente está mediada por el gen tet(B) y a la tigeciclina por una sobreexpresión de la bomba AcrAB. Las bombas de achique de las bacterias grampositivas son un poco menos efectivas para expulsar las tetraciclinas al exterior de la célula. La protección ribosomal es el mecanismo más relevante en bacterias grampositivas, aunque también existe en gramnegativas.
Principales genes implicados en los mecanismos de resistencia a tetraciclinas
| Bomba de achique (efflux) | Protección ribosomal | |
| Gramnegativos | Grampositivos | Grampositivos (gramnegativos) |
| tet(A) | tet(K) Staphylococcus | tet(O) Múltiples géneros |
| tet(B) | tet(L) | tet(S) Enterobacterias |
| tet(C) | tet(Z) | tet(M) Múltiples géneros |
| tet(D) | tet(P) Clostridium | tet(Q) Bacteroides |
| tet(E) | tet(V) Mycobacterium | tet(T) Streptococcus |
| tet(G) | tet(P) Clostridium | |
| tet(H) | ||
| tet(I) | ||
| tet(J) | ||
| tet(L) | ||
| tet(Y) | ||
La resistencia y la llegada de otros antibióticos, como quinolonas fluoradas, han desbancado gran parte de las indicaciones iniciales de las tetraciclinas. Sin embargo, siguen siendo eficaces en gran número de infecciones del adulto y han recuperado algunas indicaciones previamente desechadas por la reversión de las elevadas tasas de resistencia observadas en décadas anteriores.
Clásicamente, son de primera elección en el tratamiento de la brucelosis (habitualmente asociadas a estreptomicina) y el cólera. Son útiles en el tratamiento de infecciones por espiroquetas, en la fase inicial (eritema migrans) de la enfermedad de Lyme, en la fiebre recurrente (Borrelia spp.) y en el tratamiento y la profilaxis del paludismo causado por Plasmodium resistente a cloroquina. Se recomiendan para una gran variedad de infecciones de transmisión sexual: uretritis no gonocócica, cervicitis, linfogranuloma venéreo (Chlamydia trachomatis), sífilis en casos de alergia penicilínica y granuloma inguinal (Calymmatobacterium granulomatis). Son útiles en el tratamiento de la gastritis y la úlcera péptica asociadas a H. pylori (tratamiento combinado con otros antibióticos y un inhibidor de la bomba de protones). Se han utilizado en el tratamiento de la enfermedad de Whipple y son muy eficaces en el acné18. Podrían usarse en la profilaxis y el tratamiento del ántrax19. Aparte de otras aplicaciones no antiinfecciosas (efecto neuroprotector de minociclina)20 que aquí no es lugar para referir, las tetraciclinas de primera y segunda generación, especialmente la doxiciclina, han recuperado su original papel en las infecciones del tracto respiratorio inferior debido a la menor tasa de resistencia en neumococo y a su efecto sobre parte importante de las bacterias implicadas en la neumonía atípica.
Debido al amplio espectro de actividad, la tigeciclina está indicada en el tratamiento de las infecciones intraabdominales graves con factores de riesgo de mala evolución, donde participan con frecuencia microorganismos multirresistentes, tanto en peritonitis secundarias como en peritonitis terciarias21. También representa un tratamiento alternativo en infecciones complicadas de piel y tejidos blandos, situaciones en las que con frecuencia se encuentran implicados patógenos multirresistentes, como S. aureus resistente a meticilina, enterococo multirresistente y enterobacterias portadoras de betalactamasas de espectro ampliado10,11. Acinetobacter spp. frecuentemente muestra resistencia a un gran número de antimicrobianos, y en ocasiones, tan sólo muestra actividad frente a colimicina y tigeciclina. Por esto, el tratamiento con tigeciclina se promovió casi como el tratamiento de elección en estas infecciones. Desgraciadamente, la CMI90 de los Acinetobacter baumanii frecuentemente supera, o está en el límite, de lo que su perfil farmacodinámico indica una actuación antimicrobiana eficaz. Actualmente, no sería razonable indicar tigeciclina para infecciones por A. baumanii cuya CMI fuese superior a 1μg/ml en presencia de otros tratamientos utilizables (p. ej. imipenem sensible). Del mismo modo queda por dilucidar si las infecciones por Acinetobacter con CMI de tigeciclina de 4μg/ml pueden beneficiarse de un tratamiento con tigeciclina solo o combinado con otro antimicrobiano. La tigeciclina puede ser una buena opción terapéutica para el tratamiento de neumonías de aspiración y en un futuro próximo para infecciones complicadas por otros microorganismos emergentes como Nocardia y micobacterias atípicas.
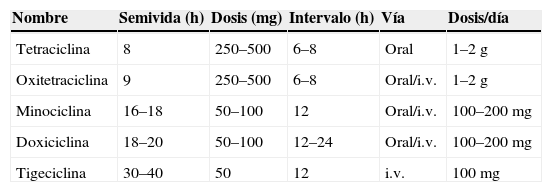
La mayoría de las tetraciclinas se deben administrar 4 veces al día para mantener concentraciones terapéuticas, pero demeclociclina y minociclina se pueden dar en 2 dosis y doxiciclina en una. La tigeciclina se administra únicamente por vía intravenosa en 2 dosis diarias (tabla 3). No está indicada la administración de tetraciclinas en niños ni en mujeres embarazadas.
Semivida, dosificación y vías de administración de las principales tetraciclinas
| Nombre | Semivida (h) | Dosis (mg) | Intervalo (h) | Vía | Dosis/día |
| Tetraciclina | 8 | 250–500 | 6–8 | Oral | 1–2g |
| Oxitetraciclina | 9 | 250–500 | 6–8 | Oral/i.v. | 1–2g |
| Minociclina | 16–18 | 50–100 | 12 | Oral/i.v. | 100–200mg |
| Doxiciclina | 18–20 | 50–100 | 12–24 | Oral/i.v. | 100–200mg |
| Tigeciclina | 30–40 | 50 | 12 | i.v. | 100mg |
i.v.: intravenosa.
Las tetraciclinas son generalmente bien toleradas y actualmente tienen relativamente pocos efectos secundarios. Antiguamente, las tetraciclinas tras dosis elevadas fueron causa frecuente de insuficiencia renal y toxicidad hepática, especialmente en mujeres embarazadas. En la actualidad están contraindicadas en estos casos, por lo que son efectos raramente observados. La doxiciclina es la tetraciclina mejor tolerada, los efectos secundarios más habituales son gastrointestinales, pero se presentan menos frecuentemente que en otras tetraciclinas. La minociclina se ha asociado con toxicidad del sistema nervioso central, especialmente trastornos vestibulares (ataxia, vértigo, tinnitus, etc.); son más comunes en mujeres, por lo que en ellas debe administrarse con precaución. Oxitetraciclina y tetraciclina presentan efectos secundarios comunes a otros compuestos de la familia.
La intolerancia gastrointestinal (náuseas, vómitos, diarreas) es el efecto secundario más importante y es dependiente de la dosis. Ésta es la reacción adversa más frecuente acaecida tras la administración de tigeciclina. Excepcionalmente, con las tetraciclinas orales se han notificado úlceras esofágicas. Para evitar estas úlceras se aconseja que al tomar la medicación se ingiera agua en abundancia y no tumbarse durante las 2h siguientes. Todas las tetraciclinas pueden producir fotosensibilidad, por lo que no debe olvidarse recomendar al paciente que limite su exposición solar. Las reacciones de hipersensibilidad son diversas (urticaria, dermatitis exfoliativa, exantema fijo medicamentoso) y suelen ser manifestaciones de fotosensibilidad; se presentan menos frecuentemente asociadas a doxiciclina y minociclina que a otras tetraciclinas; edema periorbitario y anafilaxia son raros. No es infrecuente observar micosis (oral o vaginal en mujeres) y diarrea como consecuencia de la alteración de la flora saprofita. Una consecuencia grave, aunque rara, de esta alteración de la flora es la colitis seudomembranosa. Tras la administración intravenosa pueden observarse localmente fenómenos de toxicidad tisular.
No deben administrarse tetraciclinas (excepto doxiciclina, minociclina y tigeciclina) a pacientes con insuficiencia renal previa, ya que pueden producir insuficiencia renal. Debido a su alta afinidad por el tejido óseo y dental no se recomienda su administración a embarazadas y niños menores de 8 años, ya que se depositan en dientes y huesos en desarrollo. Pueden producir decoloración de los dientes (un problema estético que no alteraría la integridad del diente) u otras alteraciones, como displasia de encías, hipoplasia dental o deformidades óseas.
Las tetraciclinas pueden interaccionar con otros fármacos: la eficacia de los contraconceptivos hormonales puede verse disminuida; los anestésicos fluorados pueden producir toxicidad renal; la fenitoína, la carbamazepina, la rifampicina y el etanol reducen la semivida de la doxiciclina. La digoxina, el metotrexato, la fenformina, las teofilinas y el litio aumentan la toxicidad de los cumarínicos. La asociación con penicilinas puede resultar antagónica. La tigeciclina puede prolongar el tiempo de coagulación, por lo que se debe monitorizar el tiempo de protrombina y el tiempo de tromboplastina parcial activado cuando este fármaco se administra junto con anticoagulantes.
SulfamidasLas sulfamidas fueron los primeros agentes antibacterianos eficaces empleados en el tratamiento de las infecciones en el hombre. Su descubrimiento durante la década de 1930 es el punto de partida del tratamiento antiinfeccioso. Son antimicrobianos sintéticos, bacteriostáticos, de amplio espectro, inicialmente con actividad frente a una gran variedad de microorganismos grampositivos y gramnegativos pero con posterior desarrollo de amplia resistencia.
Su mecanismo de acción se basa en la inhibición de la síntesis de los ácidos nucleicos bacterianos. Dentro de las sulfamidas existen numerosos compuestos con diferentes propiedades farmacocinéticas y efectos secundarios. Sin embargo, todos comparten el mismo modo de acción y es frecuente la resistencia cruzada entre ellos22–24. Actúan sinérgicamente con algunos componentes de la familia de las diaminopirimidinas, como la pirimetamina y el trimetoprima, contra bacterias y algunos protozoos. El cotrimoxazol (TMP-SXT), una asociación de trimetoprima y sulfametoxazol en proporción 1/5, es la combinación empleada más frecuentemente25. En la actualidad, el uso de sulfamidas solas es excepcional, debido a su relativa baja actividad comparada con otros antimicrobianos, al problema de la resistencia adquirida y su perfil de toxicidad. Las únicas sulfamidas de uso sistémico comercializadas en España, excluyendo alguna asociación múltiple de dudosa utilidad, son la sulfadiazina y la combinación de sulfametoxazol con trimetoprima. La combinación sulfadoxina con pirimetamina (Fansidar®) no está comercializada en España, aunque puede obtenerse a través de medicamentos extranjeros. Estos 3 preparados se encuentran en la lista de medicamentos esenciales de la Organización Mundial de la Salud7.
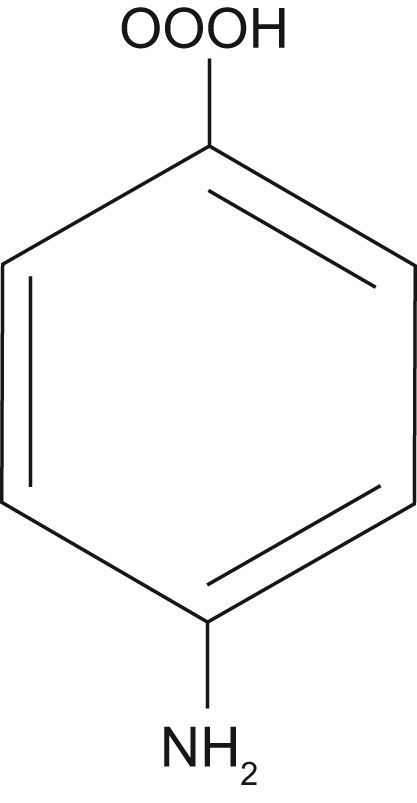
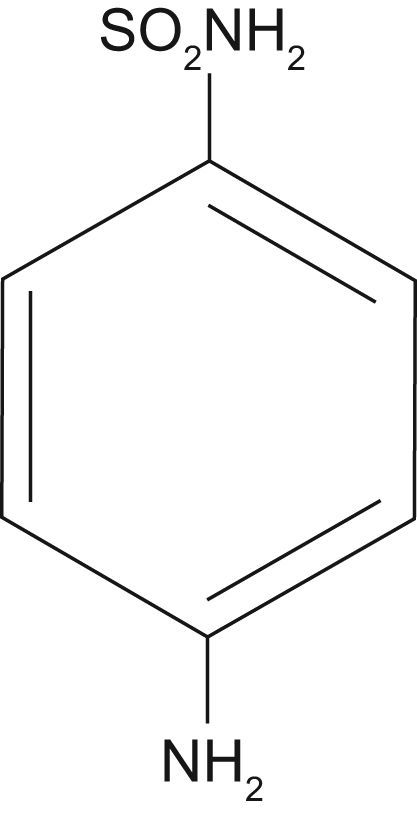
Estructura química y farmacocinéticaDerivan de la sulfanilamida. Su estructura es similar al ácido paraaminobenzoico, un factor requerido por las bacterias para la síntesis del ácido fólico (figs. 2 y 3).


Habitualmente, las sulfamidas se administran por vía oral y ocasionalmente por vía intravenosa (sulfadiazina, TMP-SXT) y tópica como la sulfadiazina argéntica. Las sulfamidas que se absorben por vía digestiva lo hacen con rapidez en el estómago e intestino delgado. En general se distribuyen bien por todo el organismo y alcanzan concentraciones cercanas al 80% de los niveles séricos en el líquido sinovial, pleural o peritoneal. La concentración en el LCR de sulfadiacina es del 40–60% de las correspondientes concentraciones plasmáticas, y la concentración del sulfametoxazol es del 80%. Se unen de modo variable y reversible a las proteínas, y los niveles alcanzados en los líquidos orgánicos están inversamente relacionados con el grado de unión a ellas. Atraviesan la barrera placentaria y alcanzan la sangre fetal y el líquido amniótico, y pueden producir efectos tóxicos. Se metabolizan en el hígado principalmente por acetilación, aunque también por glucuronoconjugación y oxidación. Los metabolitos no tienen actividad antibacteriana. La semivida de las de eliminación media (sulfametoxazol, sulfadiazina) es de 11–24h. Las sulfamidas de eliminación lenta, como sulfametoxipiridazina, tienen una semivida de 24–60h, y las sulfamidas de eliminación muy lenta, como sulfadoxina, tienen una semivida mayor de 60h.
Se eliminan principalmente por la orina, parte sin metabolizar y parte en forma de conjugados. La sulfadiacina se excreta en la orina en forma inalterada hasta un 30–44% y un 15–40% como metabolito acetilado. La excreción renal depende del pH urinario. Se une a proteínas en un 38–48%. La sulfadoxina mantiene niveles plasmáticos durante largos períodos y su concentración en la orina es muy baja. En caso de deterioro renal la dosis de las sulfamidas debe ajustarse al grado de ésta.
Espectro de actividadEstá limitado debido a la cada vez más extendida resistencia adquirida. De no considerar esta resistencia adquirida, las sulfamidas son inicialmente activas frente a un amplio grupo de bacterias grampositivas, incluyendo cepas de estreptococos, estafilococos y neumococos, aunque son naturalmente inactivas frente a Enterococcus spp. Otros microorganismos frente a los que presenta sensibilidad son Actinomyces spp., Nocardia spp., B. anthracis y Corynebacterium diphteriae. Dentro de las bacterias gramnegativas son activas a numerosas especies de enterobacterias, Neisseria spp. y patógenos respiratorios como H. influenzae, Bordetella pertussis o L. pneumophila. También son inicialmente activas frente a Yersinia pestis, Brucella spp. y algunos microorganismos involucrados en infecciones de transmisión sexual como C. trachomatis, Haemophilus ducreyi y Calymmatobacterium granulomatis. P. aeruginosa suele ser naturalmente resistente pero no S. maltophilia. Las micobacterias son resistentes, excepto algunos compuestos de larga duración con moderada actividad frente a Mycobacterium leprae. Con compuestos de la familia de las diaminopirimidinas, como pirimetamina y trimetoprima, se produce un efecto sinérgico y la combinación las hace activas frente a Toxoplasma gondii, Plasmodium spp. y Pneumocystis jirovecii.
Mecanismo de acción y resistenciasEstán estructuralmente relacionadas con ácido paraaminobenzoico y compiten con él por la enzima dihidropteroato sintasa que interviene en el metabolismo del ácido fólico. El ácido fólico es imprescindible para la síntesis de precursores de los ácidos nucleicos bacterianos. Las células de los mamíferos requieren ácido fólico preformado, ya que no pueden sintetizarlo y, por tanto, no se ven afectadas por la acción de las sulfamidas. La trimetoprima, además, inhibe otra enzima integral de las bacterias, la dihidrofolato reductasa. La actividad antibacteriana se inhibe en presencia de pus o restos de tejido necrótico (reducen la necesidad de la bacteria de sintetizar ácido fólico). Las diaminopirimidinas (como la trimetoprima), al igual que las sulfamidas, interfieren en el metabolismo del ácido fólico, por lo que combinadas tienen efecto sinérgico.
La resistencia a sulfamidas es un fenómeno creciente y generalizado, y cuando se presenta afecta a todos los componentes del grupo. Diferentes mecanismos determinan la resistencia bacteriana a las sulfamidas: disminución de la permeabilidad, expulsión activa (achique o eflujo) o alteraciones enzimáticas que por una vía alternativa o por hiperproducción permiten la síntesis del ácido fólico. La resistencia unas veces se debe a mutaciones, y otras, más frecuentemente, a la adquisición de plásmidos u otros elementos géneticos móviles que además de la resistencia a sulfamidas portan genes de resistencia a otros antibióticos. En la mayoría de las bacterias gramnegativas la resistencia a sulfamidas se debe a la adquisición de plásmidos portadores de variantes mutadas del gen DHPS23–26. A veces esta resistencia se debe a mutaciones cromosómicas. Alteraciones en este mismo gen están implicadas en la resistencia observada en cepas de Neisseria meningitidis, Plasmodium spp., S. pyogenes y S. pneumoniae, y también se han relacionado con la aparición de resistencia a TMP-SXT en cepas de P. jirovecii27,28. Hasta el momento se conocen 2 tipos de genes plasmídicos, suli y sulii, que determinan resistencia a las sulfamidas y al menos a 27 clases de plásmidos que producen 6 tipos de enzimas diferentes que confieren resistencia a trimetoprima. Con frecuencia, estos genes son elementos constantes en los integrones tipo i, el integrón encontrado con más frecuencia en cepas de casos clínicos con resistencia a múltiples antibióticos. La presencia de varios genes en un mismo elemento móvil favorece la selección de microorganismos multirresistentes, como se ha observado tras tratamientos prolongados con TMP-SXT en la profilaxis de la neumonía por P. jirovecii28.
Indicaciones clínicas y dosificaciónEl uso de sulfamidas ha disminuido según han ido apareciendo nuevos antimicrobianos más eficaces y mejor tolerados o según se ha ido incrementando el número de cepas resistentes. Su interés casi se concentra en las asociaciones con trimetoprima o pirimetamina. El TMP-SXT es primera línea de tratamiento en la infección por Nocardia spp., en la prevención y el tratamiento de la neumonía por P. jirovecii y en las enteritis por Shigella spp. Mantiene una excelente actividad frente a S. aureus, tanto sensible como resistente a meticilina, con una tasa de resistencia inferior al 5%. Este hecho junto con su comodidad de administración y el bajo coste económico hacen que represente una alternativa eficaz en el tratamiento de infecciones estafilocócicas, especialmente en el prolongado tratamiento de las infecciones óseas. Para infecciones respiratorias bacterianas actualmente es poco útil en nuestro medio, ya que el porcentaje de cepas resistentes, M. catarrhalis (>90%), H. influenzae (20–30%) o neumococo (30–50%) es muy elevado. Tampoco es una buena alternativa en la profilaxis de la infección urinaria debido a la resistencia adquirida (el 20–35% en E. coli y resistencia natural en Enterococcus spp.), aunque constituye una excelente opción terapéutica para el tratamiento en presencia de un antibiograma que muestre sensibilidad a su agente causal. El TMP-SXT también está indicado en el tratamiento de algunos parásitos intracelulares de hábitat intestinal como Isospora belli o Cyclospora cayetanensis. La sulfadiazina asociada a pirimetamina es el tratamiento de elección en la toxoplasmosis del niño y del adulto, incluido los infectados por el virus de la inmunodeficiencia humana. La sulfadiazina argéntica es útil en quemaduras y úlceras por decúbito en segundo y tercer grado.
Efectos adversos y contraindicacionesUna de las principales desventajas de las sulfamidas en comparación con otros antimicrobianos más recientemente comercializados es la elevada frecuencia de efectos secundarios. Las reacciones de hipersensibilidad son frecuentes: exantema, fiebre, anafilaxia, eritema multiforme, dermatitis necrosante, síndrome de Stevens-Johnson (raro pero a menudo grave). Pueden producir trastornos digestivos como náuseas, vómitos y diarrea. Los trastornos hepáticos son raros. También pueden producir alteraciones hematológicas (anemia hemolítica en pacientes con déficit de glucosa-6-fosfato deshidrogenasa, anemia megaloblástica, por su acción antifólica, etc.). Están contraindicadas en el último trimestre de embarazo porque pueden desencadenar kernicterus. No se recomiendan durante la lactancia ni en los primeros meses de vida.
Pueden producir interacciones medicamentosas: potencian a anticoagulantes e hipoglucemiantes orales, metotrexato, diuréticos tiazídicos, fenitoína y uricosúricos. Son potenciadas por indometacina, fenilbutazona, salicilatos y probenecid.
MetronidazolEl metronidazol es un compuesto 5-nitro-imidazol introducido en el año 1959 para el tratamiento de infecciones producidas por Trichomonas vaginalis. Su potente actividad anaerobicida lo ha convertido en un referente y el comparador obligado (gold standard) para estimar la actividad relativa de cualquier fármaco con actividad frente a anaerobios29–32. Es útil en muchas infecciones parasitarias, aunque nuevos compuestos (p. ej.: tinidazol, nitazoxanida, etc.) lo han desbancado parcialmente.
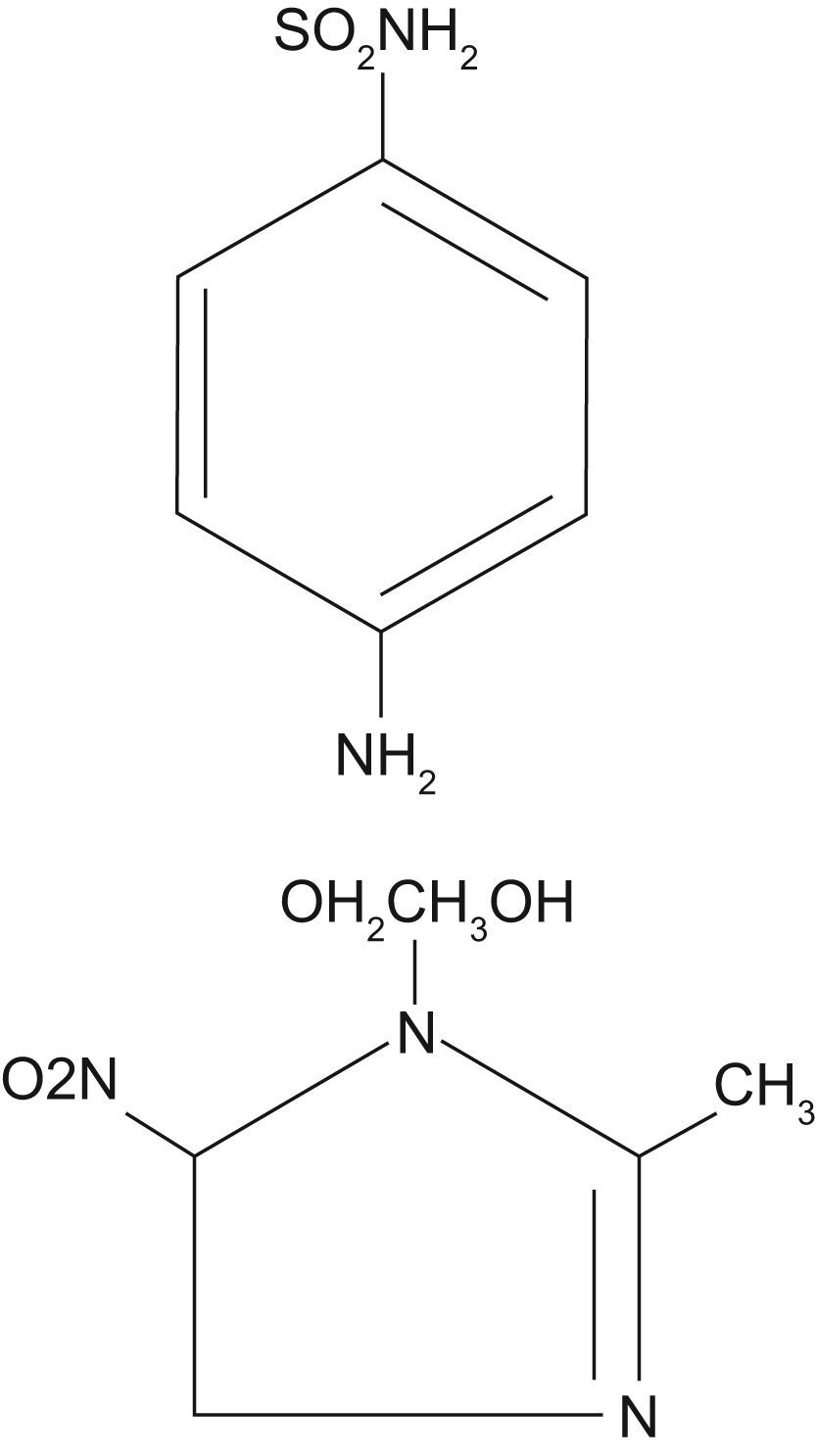
Estructura química y farmacocinéticaAdemás del metronidazol, se han desarrollado otros 5-nitroimidazoles de características farmacocinéticas y antimicrobianas similares (fig. 4) (tinidazol, ornidazol. benznidazol, etc.). De todos estos compuestos, tan sólo metronidazol, tinidazol y ornidazol están comercializados en España. Ornidazol sólo se encuentra en solución alcohólica para tratamiento parenteral y se utiliza poco, y tinidazol, comercializado en comprimidos, suele utilizarse en tratamientos con dosis única.

El metronidazol, el ornidazol y el tinidazol tienen propiedades farmacocinéticas similares. Se absorben muy bien por vía oral y su biodisponibilidad es superior al 90%. Las concentraciones máximas se observan entre 1 y 2h después de su administración y son proporcionales a la dosis (250 o 500mg o 2g vía oral de metronidazol producen concentraciones séricas máximas de 6, 12 y 40μg/ml, respectivamente); en pacientes con tratamiento intravenoso, tras una dosis inicial de 15mg/kg seguidos de 7,5mg/kg cada 6h, las concentraciones pico-valle son de 25–18μg/ml. Los alimentos no disminuyen su absorción pero pueden retrasarla. La biodisponibilidad del metronidazol en supositorios es del 60–80%, y tras su administración vaginal es del 20%. La semivida del metronidazol es de 6–12h y algo mayor en el caso del tinidazol y el ornidazol.
El metronidazol se distribuye ampliamente y alcanza todos los tejidos y líquidos por vía oral o intravenosa (saliva, bilis, huesos, hígado, pulmón, líquido peritoneal, semen y secreciones vaginales). Se une escasamente a proteínas (menos del 20%). Atraviesa la placenta y la barrera hematoencefálica. En el LCR alcanza niveles terapéuticos incluso sin inflamación meníngea. Se excreta en la leche materna, por lo que no se recomienda en madres lactantes. Se metaboliza en el hígado en un 30–60% de la dosis. El metabolito principal es el 2-hidroximetil metronidazol, que tiene cierta actividad antibacteriana y antiprotozoaria.
La eliminación es fundamentalmente a través de la vía renal. Cerca del 60–80% se excreta en la orina (el 20% como fármaco sin cambios) y el 6–15% en las heces sin metabolizar. Se elimina rápidamente por hemodiálisis, pero no por diálisis peritoneal. La disminución en la función renal no altera la eliminación, pero si el paciente está anúrico es preferible evitar dosis elevadas debido a la posibilidad de la acumulación de sus metabolitos. En pacientes con deterioro de la función hepática puede disminuir su eliminación, por lo que si la insuficiencia es grave será preciso modificar la dosis o la frecuencia de administración.
Espectro de actividad, mecanismo de acción y resistenciasEl espectro de actividad incluye protozoos, bacterias anaerobias y algunas microaerófilas (no activo frente a Actinomyces). Es muy activo frente a prácticamente todo tipo de bacilos gramnegativos anaerobios, frente a Gardnerella vaginalis y H. pylori. Su espectro antiprotozoario incluye E. histolytica, Giardia lamblia, T. vaginalis y B. coli.
Ejerce su acción antibacteriana y antiprotozoaria por desestructuración del ADN. Tras ingresar en la célula mediante difusión pasiva, proteínas del metabolismo anaerobio (proteínas de transporte de electrones de bajo potencial redox) reducen químicamente al metronidazol. Estas proteínas son exclusivas de algunos parásitos y de bacterias anaerobias y algunas microaerófilas. El metronidazol reducido produce pérdida de la estructura helicoidal del ADN, rotura de la cadena e inhibición de la síntesis de ácidos nucleicos y muerte celular, y genera compuestos que son tóxicos para la célula33. El metronidazol es mucho más activo cuando actúa en lugares donde existe anaerobiosis. Además de sus propiedades antimicrobianas, se le atribuye un efecto antiinflamatorio, antioxidante e inmunomodulador.
El principal mecanismo de resistencia es por alteración de las enzimas implicadas en la activación intracelular del fármaco, necesaria para la producción de sus metabolitos activos34–36. La resistencia adquirida en bacilos gramnegativos anaerobios es infrecuente y no se ha detectado aumento en los últimos años (<1% de cepas de Bacteroides grupo fragilis son resistentes en nuestro medio37,38. El metronidazol es muy activo frente a Clostridium difficile, aunque su utilidad para tratar las diarreas u otro tipo de afecciones por este microorganismo se cuestiona actualmente debido a la facilidad con que se seleccionan subpoblaciones resistentes, especialmente al no utilizar dosis elevadas39. La resistencia en H. pylori se sitúa entre el 25–50%.
Indicaciones clínicas y dosificaciónEs eficaz en el tratamiento de la mayoría de las infecciones por anaerobios. Es útil en combinación con aminoglucósidos en el tratamiento de infecciones polimicrobianas de tejidos blandos e infecciones mixtas aerobias-anaerobias intraabdominales y pélvicas. Está indicado en asociación con otros antibióticos en el tratamiento de los abscesos cerebrales de origen sinusal, dental, ótico, pulmonar o criptogenético, en los que hay que sospechar la presencia de bacterias anaerobias. También está indicado en endocarditis infecciosas por gérmenes anaerobios. Anteriormente se ha mencionado su limitación en el tratamiento de la colitis postantibiótica por C. difficile. En infecciones mixtas en donde se presuponga la presencia de estreptococos anaerobios o facultativos se aconseja incluir una penicilina o clindamicina, ya que es poco o nada activo frente a ellos. Es uno de los antibióticos más activos de los utilizados en el tratamiento de la infección por H. pylori; su única desventaja es el elevado porcentaje de cepas resistentes. Se lo sigue considerando de primera línea en el tratamiento de vaginosis por G. vaginalis, tricomoniasis y giardiasis.
La dosis de metronidazol más habitual en el adulto es de 500mg/8h, sea oral o intravenoso, pero hay variaciones en sus indicaciones. Para la tricomoniasis fuera y dentro del embarazo y como profilaxis ante un caso de violación se recomienda una dosis única de 2g40; para la vaginosis, 500mg/12h oral×7 días; para giardiasis, 250mg/8h oral×5–7 días. En el niño, las dosis oscilan entre 15–50mg/kg/día (sin superar los 750mg/día), oral o intravenosa, y se recomienda administrarlo en 3 dosis.
Efectos adversosPor lo general, el metronidazol y los otros 2 5-nitroimidazoles comercializados son bien tolerados. Las reacciones adversas más frecuentes son náuseas y diarrea. Algo menos frecuentes son mareos, dolor de cabeza, pérdida del apetito, vómitos, dolor o calambres abdominales. Aunque mucho más raro, también pueden presentarse cambios en la sensación del gusto, estreñimiento, sequedad bucal, glositis, estomatitis, cefalea, pigmentación oscura de la orina, flebitis en el sitio de la inyección venosa, leucocitopenia leve y reversible, trombocitopenia, prurito, erupción, insomnio, artralgias, fiebre. Otras reacciones graves del metronidazol y el ornidazol se pueden ver en enfermos que reciben altas dosis o tratamientos prolongados; entre ellas, destaca la polineuritis sensitiva y algunos efectos sobre el sistema nervioso central: incoordinación, ataxia, confusión, irritabilidad, depresión, abatimiento o insomnio. La presentación de estos signos obliga a interrumpir su administración.
El metronidazol y el tinidazol pueden producir reacciones tipo disulfiram cuando se administra a pacientes que ingieran alcohol. El metronidazol inhibe el metabolismo y aumenta el nivel sérico de la fenitoína y anticoagulantes orales, carbamacepina y ciclosporina. Aumenta la toxicidad del litio, el fluorouracilo y la cloroquina. Puede reducir la eficacia de los anticonceptivos orales. Los barbitúricos y los corticoides aumentan el metabolismo hepático y la cimetidina lo reduce.

