





Introduction
Highly active antiretroviral therapy (HAART) was a revolution in the management of HIV-infected patients, drastically reducing mortality and the incidence of opportunistic infections. In spite of the undeniable clinical benefits obtained, between 20% and 50% of patients who initiate HAART present virological failure during the first year of treatment and the incidence of failure increases rapidly during successive treatments1-5. Treatment can fail for many reasons, which may be patient-, virus-, or drug-related6. There can be no doubt as to the importance of obtaining a suitable virological response at the focus of virological multiplication. If the concentration is greater than needed to inhibit the virus, viral replication will be suppressed and development of resistance mutations will be avoided. If the concentration is low, selective pressure on the virus will lead to the development of resistance to one or more drugs and virological failure. If, on the contrary, the concentration of the drug is very high, toxicity could increase. At present, pharmacological toxicity is the main reason for suspending treatment and an important contributor to poor adherence.
Monitoring the plasma concentration of some drugs has been in use for more than 30 years. The introduction of this scientific discipline in normal clinical practice has enabled us to reduce morbidity, mortality, adverse effects and hospital stay among some patients, although it can only be applied to a small group of drugs such as oral anticoagulants, digoxin, theophyllines, aminoglycosides, vancomycin, cyclosporine and other immunosuppressors or some anti-epilepsy drugs. HIV therapy uses fixed doses of drugs and therapeutic drug monitoring does not form part of normal clinical practice, even though ART have certain characteristics which make them appropriate for therapeutic drug monitoring. The value of adjusting the antiretrovirals dose to maintain specific concentrations remains controversial. In recent years, therapeutic drug monitoring has aroused both interest and speculation concerning its role in the optimisation of ART7-13.
The concept of therapeutic drug monitoring
This strategy allows the dose of a drug to be modified according to its plasma concentration, in order to maintain it within previously defined therapeutic limits, with the objective of improving therapeutic efficacy and/or avoiding toxicity.
Necessary requirements for drug monitoring to be potentially useful
Determination of the drug plasma concentration
Liquid chromatography (HPLC) techniques enable us to precisely determine the plasma or serum concentrations of all the currently available PIs and NNRTIs14-16. Furthermore, we can simultaneously determine the concentration of several drugs in a single chromatography. Despite the fact that this technique has only been applied in a few large hospitals and reference centres, it can be carried out in any clinical laboratory with a chromatograph and qualified staff. At present, all laboratories can participate in an international quality control programme for the determination of PI and NNRTI concentrations17,18. The cost of determining the concentration of a drug is considerably lower than that of other routine tests in HIV-infected patients. For example, determining the Cmin of a PI or an NNRTI in an external laboratory costs 20-30 €.
Determination of the NRTI plasma concentration is equally feasible. Nevertheless, given that they need to be phosphorylated inside the cells to exercise their action, the correlation between the blood levels of most compounds and their antiretroviral activities and/or potential toxicity is poor. Consequently, NRTIs are not currently recommended for therapeutic drug monitoring, although the interest in monitoring NRTI levels has shown us that tenofovir increases the plasma concentration of didanosine by 40%-60%. This information has enabled us to recommend reducing the dose of didanosine in patients who receive concomitant tenofovir.
Wide interindividual variability
of pharmacokinetic parameters
Therapeutic drug monitoring is only meaningful when the administration of the same dose to different patients can lead to variable concentrations. Were this not the case, the concentration of the drug would be very predictable, and knowing the dose would almost be the same as knowing the plasma concentration.
At present, patients treated with PIs or NNRTIs receive the same dose, and one of the most common problems is the huge interpatient variability of the plasma concentrations19-27. The AUC, Cmax or Cmin frequently vary more than 10-fold between different patients receiving the same dose. This wide interpatient variability is basically due to differences in the activity of the isoenzymes of the cytochrome P450 system and the transporter proteins or efflux pumps (glycoprotein P, etc.) responsible for the absorption and metabolism of PIs and NNRTIs28-32. Other factors, such as weight, age, presence of chronic liver disease and drug-drug interactions can also play an important role.
Even when small doses of ritonavir are administered to inhibit cytochrome P450 and the efflux pumps, interpatient variability is still very wide33-35.
Scarce intrapatient variability of pharmacokinetic parameters
For therapeutic drug monitoring to be viable, it is important that a single determination of the plasma concentration has a considerable value. If there was a wide intrapatient variability, monitoring would be practically impossible, as determining the plasma concentration of a drug today would not provide us with information on the plasma concentration tomorrow, next week or next month.
Few studies have evaluated the intrapatient variability of ART, but those that do have observed that the concentration of PIs and NNRTIs in the same patient is relatively constant, with variation coefficients of 30-45%21-23,26,36. Therefore, a single determination of these drugs would give us reliable information on the concentrations maintained by the patient. Obviously, pharmacokinetic stability will break down if patient circumstances change, especially if adherence is poor, but also if absorption is altered (due to diarrhoea, for example, or intestinal malabsorption) or if the patient takes other drugs or non-pharmacological products which interact with antiretrovirals.
Good correlation between plasma concentration and therapeutic efficacy or toxicity
If the correlation between plasma concentration and clinical response was not good, therapeutic drug monitoring would be of no use. This seems to be the case with NRTIs. No clear relationship has been established between the plasma concentration of NRTIs and response to therapy. NRTIs are prodrugs which are activated inside the cell by kinase action and become active triphosphate derivatives. These reactions limit the amount of intracellular active drug. Determination of the intracellular concentration of NRTI-triphosphate could be more useful for predicting the response to therapy37, but this requires very complex techniques. Mass spectrometry is used, although it is only available in some research laboratories.
Unlike NRTIs, PIs and NNRTIs do not require intracellular metabolization to be active. Several studies show the relationship between PI plasma concentration and response to therapy, especially in ART-naive patients38-59. A relationship has also been observed between high concentrations of ritonavir60, indinavir61-65, nelfinavir66 and amprenavir56, and a greater incidence of adverse reactions. Some types of digestive toxicity from PIs are caused by local action on mucosa and it is no surprise that the plasma concentration of nelfinavir is not associated with diarrhoea67.
As far as NNRTIs are concerned, both efavirenz and nevirapine have a longer elimination half-life than PIs. This is seen through smaller oscillations in plasma concentrations during this phase and the possibility of using a single sample four hours after taking nevirapine when it is administered every 12 hours, or after 12 hours, both for nevirapine and for efavirenz, when they are administered once daily, to predict the Cmin68-70. Nevertheless, interpatient variations in plasma concentrations are as marked as with PIs23-26,69-71, and there are data to support a relationship between the levels of both drugs and their efficacy, although the minimum concentrations associated with long-term efficacy have been neither clinically validated nor clearly defined23,24,26,72-74. It may be very important to discover cases with low concentrations of nevirapine or efavirenz given the speed with which resistance can develop, since a single mutation conditions the loss of efficacy. The toxicity of NNRTIs has also been related to plasma concentrations. High concentrations of efavirenz have been associated with more frequent CNS alterations23,32,75,76, although some studies have not observed this association77. Similarly, high concentrations of nevirapine have been associated with liver toxicity78 and rash79.
If we take into account the significant correlation between pharmacokinetic parameters such as AUC, Cmax and/or Cmin and the response to ART or toxicity, it seems logical to suppose that maintaining plasma concentrations within certain levels will optimise its use.
Narrow therapeutic margin
Drugs with a wide therapeutic margin can be administered relatively easily at a fixed dose, and effective, non-toxic concentrations can be reached. Therapeutic drug monitoring will be more useful when the difference between the effective concentration and the toxic concentration is small. Most PIs have a relatively small therapeutic margin and the mean Cmin of patients receiving non-boosted PIs is slightly above the effective concentrations. Around 30-40% of patients have been estimated to present PI concentrations lower than those which are considered effective80,81. The administration of small doses of ritonavir enormously increases the plasma concentrations of PIs and, at present, all PIs, with the exception of nelfinavir, are usually combined with ritonavir33,82-84. It is extremely unusual for the concentration of saquinavir85, indinavir34, amprenavir86 or lopinavir87 boosted with ritonavir to be sub-therapeutic in patients who do not present resistance to PIs, except in cases of poor adherence. Nevertheless, therapeutic drug monitoring of PIs boosted with ritonavir is still useful, given that wide interpatient variability is maintained and that, by increasing the concentration of the PI, the number of adverse effects can rise. In addition, even if the concentration is therapeutic for non-resistant viruses, it may be insufficient for viruses with several mutations.
Characteristics which make the monitoring of antiretrovirals difficult
Confusion between adherence to therapy and plasma concentration
Adherence to ART is essential if therapy is to be suitably efficacious and durable, although it is difficult to maintain good adherence to all drugs, if we take into account doses, dosing intervals and dietary requirements. For therapeutic drug monitoring to be clinically useful, a knowledge of adherence is necessary and adherence must be good. A suitable plasma concentration does not necessarily mean that adherence will be good, as the patient may be more attentive to taking medication before the monitoring visit. A low plasma concentration may reflect a pharmacokinetic problem, poor adherence, or both. It is therefore important to evaluate and optimise adherence88 and, if possible, determine the drug plasma concentration after ingestion. If the concentration is low, we should insist on the importance of adherence, as measuring adherence can be difficult in some patients. In this context, sub-therapeutic concentrations can help us to discover cases of poor adherence. The plasma concentration is more variable in patients with poor adherence88, and sub-therapeutic concentrations of PIs or NNRTIs have been used as another parameter to be taken into account when evaluating adherence89-92. Therapeutic drug monitoring is of little use to know the adherence of drugs with a short half-life, but it may be more useful in the case of drugs with a longer half-life, such as NNRTIs or some boosted PIs (e.g. lopinavir/ritonavir). If adherence is correct, we should rule out other causes, such as pharmacokinetic reactions or intestinal malabsorption, which can lead to sub-therapeutic concentrations.
Difficulties in establishing an efficacious concentration
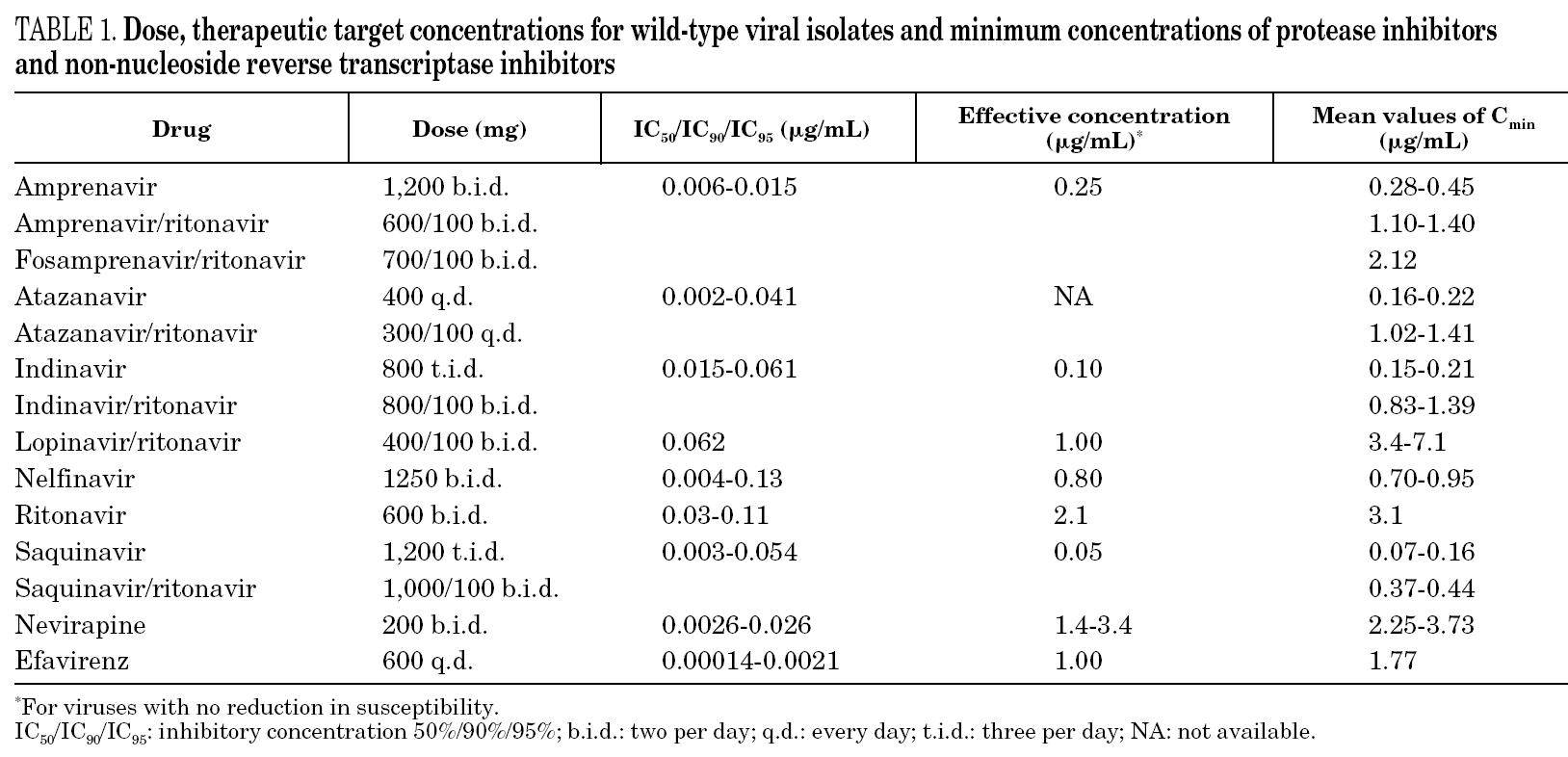
Despite abundant literature showing the relationship between the concentration of a drug and response to therapy, the therapeutic range of PIs and NNRTIs in different situations is not clearly defined. Table 1 shows the proposed therapeutic concentrations in some studies, although we should no forget that these data have not yet been sufficiently validated in clinical practice.

The first doubt concerns which of the pharmacokinetic parameters (AUC, Cmax or Cmin) best predicts the clinical efficacy of PIs or NNRTIs. These parameters are closely related, even though they provide us with different pharmacokinetic data.
The AUC tells us about the total exposure to the drug and has been related to efficacy and toxicity48,51,65,85. The main disadvantage is that, to calculate the AUC, a large number of blood samples are necessary throughout the dosing interval (8, 12 or 24 hours), with the result that determination is much more complex than with isolated concentrations, both for the patient and healthcare personnel.
The Cmax tells us the maximum exposure to the drug and is essentially associated with toxicity56,60-62,65,85, although also with virological efficacy. In a recent study, the Cmax of indinavir was the only parameter associated with an increase in the number of CD4 lymphocytes in patients with an undetectable viral load, and this suggests a pharmacokinetic-pharmacodynamic relationship which is different with respect to the effect on immune reconstitution and virological efficacy52. The problem with determining Cmax is that the point at which it is reached after ingestion varies a great deal from patient to patient, so that, to detect it correctly, it is necessary to take several blood samples during the first hours after ingestion.
The Cmin tells us the lowest plasma concentration during the dosing interval. The relative ease of determining this parameter is a considerable advantage, as only one blood sample is necessary just before the next dose is taken. For HIV replication to be inhibited permanently, effective concentrations (EC) of the drug during the therapeutic interval are necessary, so that, if the Cmin is greater than the EC, the virus would be inhibited correctly. This alleged EC is a theoretical concentration which can be found in the literature and which has been calculated in vitro from a phenotypic study of strains susceptible to the virus. In this way, we can determine the concentration of the drug necessary to inhibit 50% (IC50), 90% (IC90), or 95% (IC95) of viral replication. This inhibitory concentration is corrected by the drug's binding to plasma proteins to obtain a parameter which would theoretically correspond to the EC (EC50, EC90, EC95). This correction by binding to proteins can occur in several ways, with very different results12. Since viral replication must be inhibited as much as possible, the EC95 or the EC90 would be more suitable than the EC50, although the EC50 is generally used as it can be determined more precisely.
The Cmin is the most widely studied pharmacokinetic parameter in the literature, and that which is most associated with virological efficacy48,50,56,65,85,93,94, although it is also associated with toxicity60,63. Cmin is probably the most useful parameter with regard to cost-benefit relationship for the therapeutic drug monitoring of PIs, although it does have three disadvantages. The first is that for some PIs, the concentration continues falling for some time after the drug has been taken, until intestinal absorption reverts the trend. For this reason, a difference is made between the pre-dose, or trough, concentration (Ctrough), which is usually determined, and the minimum concentration (Cmin), which is the lowest concentration. These terms are often confused, and the literature generally refers to Cmin when it means Ctrough. For most drugs, both concentrations are very similar, as the concentration begins to increase shortly after administration, and the terminological confusion is not clinically relevant. For other drugs, such as lopinavir or ritonavir, the difference can be considerable, since the concentration continues to fall for one or two hours95. Fortunately, both the Ctrough and Cmin of lopinavir are much higher than that which is necessary to inhibit sensitive viruses, but when the virus has accumulated a certain number of resistance mutations to PIs, this difference could be important. A second disadvantage of Cmin is the circadian rhythm of some PIs, such as nelfinavir or saquinavir, with concentrations which are considerably lower at night than in the morning96. In these cases, the lowest concentration during the dosing interval would be the night-time Cmin. A third problem is that Cmin, unlike Cmax and AUC, is usually determined without directly observing the administration of the previous dose of the drug. It is therefore difficult to take blood samples at the expected time (8, 12 or 24 hours after intake, depending on the dosing interval), because the patient has taken the drug early/late, or because he/she arrived late for the visit or because of the workload of the person taking the sample. With PIs, a poor choice of extraction time can provide a sample which is not representative of what we wish to evaluate. NNRTIs, in particular efavirenz, have a prolonged half-life, so that lack of precision with sampling time has less effect on the results. In the study by Marzolini et al23, where the concentration of efavirenz was associated with therapeutic efficacy and toxicity, samples were taken indiscriminately between eight and twenty hours after administration.
Data from the literature have enabled us to generate concentration-time curves which include the percentiles 10, 25, 50, 75 and 90 of the simulated concentrations of the main therapeutic regimens with boosted and non-boosted PIs and with NNRTIs97. These curves allow us to extrapolate, with acceptable precision, the result of a sample obtained at any time interval to the population parameters. This method requires an accurate knowledge of the time of administration and the time of sampling. For drugs with a short half-life, as is the case with most PIs, errors of just a few hours can render the result inaccurate. For drugs with a prolonged half-life, such as the NNRTIs, this error is of little importance.
An additional problem when determining whether the concentration lies within therapeutic margins is that we must realize that what we are determining is the total concentration of the drug and not the free fraction, i.e. the part which spreads to the tissues and inhibits the virus. Both for PIs and for NNRTIs, the greater part of the drug in plasma is bound to plasma proteins, mainly to acid alpha1-glycoprotein and albumen. Alpha 1-glycoprotein is an acute phase reactant and its concentrations can vary considerably under certain circumstances12, for example, in the case of intercurrent infections. Therefore, it may be important to determine the concentration of a drug in a stable clinical situation. There is noticeable interpatient and intrapatient variability in the protein-bound fraction of indinavir98, and changes have been observed in the proportion of free lopinavir during the therapeutic interval99, although the clinical significance of this is unknown.
The presence of active metabolites which contribute to the overall efficacy of drugs can also make it difficult to interpret the plasma concentration. Nelfinavir is the only drug susceptible to therapeutic drug monitoring for which an active metabolite (M8) is known, with measurable concentrations in plasma. M8 is as active as nelfinavir itself and is less bound to plasma proteins. The precise role it plays in the overall activity of nelfinavir is unknown. Co-administration of ritonavir and nelfinavir slightly increases the concentration of nelfinavir but greatly increases the M8/nelfinavir quotient100,101.
Finally, we must not forget that the necessary concentration of the medication must be interpreted on an individual basis. Experienced patients, or those who are infected by viruses with mutations which reduce susceptibility to PIs, may require higher concentrations of PIs (see section "Inhibitory quotient). Concomitant medication will also play an important role, as this can be more or less potent and show synergy with the drug we are monitoring.
All these difficulties mean that considerable pharmacokinetic and pharmacodynamic knowledge is necessary in order to interpret the plasma concentration of a drug and provide a suitable dose adjustment.
Results of clinical trials
For monitoring of antiretrovirals to be applied to clinical work, it is essential to have the results of randomized studies which validate this therapeutic strategy. Several trials have been, or are being, carried out both at the beginning of treatment (to prevent the risk of virological failure or to avoid adverse effects), and in patients with previous failures (to adapt the concentration of the drug to the resistance of the virus).
The first study102,103 included 40 naive patients who began therapy with zidovudine, lamivudine and indinavir (800 mg/8 h), and who were randomized to receive the standard doses or to undergo therapeutic drug monitoring of the three drugs. Seven patients abandoned treatment early and were excluded. In the monitoring group, doses were adjusted at week 4 using the plasma concentration from week 2. The dose of zidovudine, lamivudine and indinavir was modified in 44, 31 and 81% of patients, respectively. The therapeutic concentrations defined a priori were reached in 14 of the 16 patients monitored and in 3 of the 17 who received the standard dose. After one year of follow-up, the number of patients with a viral load < 50 copies/mL was greater in the monitored patients (15 out of 16 compared with 9 out of 17). Although the number of patients studied is small, the differences are significant in favour of the therapeutic drug monitoring group.
The ATHENA study104 included 147 naive patients who began a regimen with nelfinavir (n = 92) or indinavir (n = 55) and who were randomized to receive therapeutic drug monitoring or not. In both groups, the PI concentration was determined and, in the monitoring group, the result was reported to the attending physician, together with a recommendation to adjust the dose if necessary, whereas in the control group the result was not reported. The drug concentration was expressed as the quotient of the value obtained in a sample taken at any time after administration and the population reference value of a concentration-time curve. For nelfinavir, the dose was increased if the quotient was below 0.9, whereas for indinavir, it was increased or reduced if the quotient was not within the range 0.75-2. After one year of follow-up, the number of patients with a viral load < 500 copies/mL by intention-to-treat (loss = failure) was significantly greater in the monitoring group than in the control group (78.2% and 55.1%). Similarly, a lower number of patients interrupted treatment in the monitoring group (17.4% and 39.7%). In the group treated with nelfinavir, the best outcome of the monitoring group was due mainly to a lower number of virological failures (2.4% and 17.6), whereas in patients treated with indinavir it was due to fewer toxicity-induced changes in therapy (14.3% and 29.6%). The authors conclude that therapeutic drug monitoring improves response to therapy.
In the PharmaAdapt study105,106, patients with virological failure received rescue therapy adapted to the genotype of the virus and were randomized to compare follow-up with therapeutic drug monitoring and standard follow-up. The study included 257 patients and the results of the 183 who received PIs have been published. A trough concentration greater than the IC50 corrected by the binding of plasma proteins described in the literature for non-resistant viruses was considered therapeutic. In the therapeutic drug monitoring group, the PI dose was modified at week 8 in 23.5% of patients based on the Cmin of week 4. At week 12, and at week 32, no differences were observed in viral load or in the number of patients with an undetectable viral load between both groups. Nevertheless, it cannot be concluded that therapeutic drug monitoring is not useful because the results of the study may be invalidated by several methodological problems. First, the choice of therapeutic concentrations, which, with some reservations, could be adequate in naive patients, but not in patients with previous virological failure. Furthermore, the dose adjustment is not made until week 8, which seems to be long enough for new mutations to appear.
The GENOPHAR study107 has not yet been published and we only have data from a congress communication. In this study, 134 patients with virological failure were randomized by comparing the efficacy of treatment adapted only to the genotype with that which was adapted to the genotype and with therapeutic drug monitoring. Doses were modified at week 8 in 20% of patients and the results were analysed at week 12. No significant differences were observed with or without therapeutic drug monitoring. This study has considerable problems in its design and the interpretation of data, some of which can be superimposed on the PharmaAdapt study. In addition, the criteria used to adjust the dose are not clear, and the analysis is carried out 4 weeks after adjusting the dose.
Selection of patients for whom therapeutic drug monitoring can be particularly useful
Current efficacy of ART is high, and as many as 80-85% of patients with good adherence have an undetectable viral load after one year of treatment. With these figures, the contribution of universal therapeutic drug monitoring may not be very important. Unless it is also used as another measure of adherence, at most it could improve the efficacy of ART by 5%. Given these limitations, the cost and difficulty of implementing therapeutic drug monitoring, it is clear that it cannot be applied easily to all patients. On the other hand, patients with a special risk of presenting sub-inhibitory or extremely high concentrations may obtain a considerable benefit.
In the first place, monitoring is recommended when there are likely to be pharmacokinetic interactions. The amount of possible interactions in HIV-infected patients receiving ART is enormous12,28, depending mainly on the inhibition or induction of the enzyme systems responsible for the absorption or metabolism of the medication (cytochrome P 450, transporter proteins). In general, PIs are enzyme inhibitors and NNRTIs are enzyme inducers. Many other drugs may be important enzyme inducers (e.g. rifampicin, rifabutin) or inhibitors (e.g. azoles). In tuberculosis, which is often associated with HIV infection, therapeutic drug monitoring can be recommended for some drugs, due to the drug-drug interactions which the disease leads to108-111. Even the so-called "natural medicine" can cause interactions with ART112. Sometimes, combinations of inhibitors and/or inducers can give rise to very complex and unpredictable interactions, as has recently been observed with the combination of lopinavir/ritonavir and amprenavir, which lead to marked drops in the concentrations both of lopinavir and amprenavir113-114. Therefore, therapeutic drug monitoring is essential in these cases. There are many guidelines for dosing drugs with known interactions, but we must not forget that the recommendations are generally based on mean concentrations. The wide interpatient variability means that, in many cases, the concentrations obtained are sub-therapeutic or extremely high. In the case of interactions, if possible, it would be advisable to know the concentration of antiretrovirals before and after adding the new drug in order to evaluate the magnitude of the interaction and modify the dose accordingly.
Another situation where therapeutic drug monitoring could prove useful is the appearance of adverse effects. It is sometimes difficult to know which drug is responsible for a particular toxicity, but many adverse effects are clearly associated with specific drugs and a dose reduction could revert, for example, the renal toxicity of indinavir or the toxicity of other PIs60,61.
Therapeutic drug monitoring could also be useful in once-daily regimens with drugs which do not have a long half-life, such as PIs boosted with ritonavir. The Cmin of most PIs boosted with ritonavir and of nevirapine is lower if the same dose of the once-daily drug is spread over two intakes69,85,87,115-119. It is important to know the Cmin to avoid exposure to sub-inhibitory concentrations, especially when there already exists a certain degree of resistance to the drug.
Therapeutic drug monitoring is particularly important in the case of virological failure, which may be associated with a sub-therapeutic concentration of the drug. In some cases, increasing the concentration (e.g. PI boosted with ritonavir) could make the drug efficacious again120,121. The therapeutic efficacy of successive regimens after virological failure is increasingly low. In patients with few therapeutic options, ART must be maximized to give an acceptable response, and correct pharmacological concentrations will enable us to increase virological efficacy and reduce toxicity (see Section "Inhibitory quotient").
In patients with chronic liver disease, there have been reports of greater variability122 or higher concentrations of some drugs123,124, which can increase toxicity125. In this sense, therapeutic drug monitoring may be especially useful in cases of advanced liver disease to attempt to reduce toxicity in patients who are already prone to complications.
Some covariables, such as weight, sex or race can modify the pharmacokinetics of antiretrovirals. Overall, the correlation between concentration and weight is not very good in adults and neither PIs nor NNRTIs are adjusted to weight, although in overweight patients there may be a noticeable influence. In extremely overweight patients, it may be useful to determine the concentration in order to correct sub-therapeutic concentrations, and in very underweight patients, monitoring may help avoid toxicity. There have been reports in women of high of some drugs such as saquinavir, regardless of weight or body mass95,126. This greater concentration in women has been associated with greater efficacy, but also with greater toxicity, so that if toxicity appears, it may be useful to determine the concentration and reduce the dose if it is high. The risk of presenting adverse reactions to nevirapine (skin rash and liver toxicity) is considerably higher in women than in men127. Special mention must be made of the racial differences related to polymorphisms at codon 516, which may be responsible for higher levels of efavirenz-induced toxicity. This finding guarantees the possibility of pharmacogenomic studies alongside pharmacokinetic studies to optimise treatment.
Pregnant women are a population in which therapeutic drug monitoring is important128,129. It has been shown that pregnancy can modify the pharmacokinetics of some drugs, especially during the third term, with wide variability in plasma concentrations130-134. The concentrations of non-boosted indinavir and saquinavir sometimes fall to sub-therapeutic values, whereas that of nelfinavir is hardly modified. If we determine the Cmin during the first term, we can have a reference of the baseline concentration. If it falls during the second or third term, the dose can be adjusted to avoid sub-therapeutic concentrations.
Therapeutic drug monitoring can be particularly useful in children135. Despite the fact that we have more results from pharmacokinetic studies on children41,130,136-142, many antiretrovirals have not been studied in neonates or small children. The pharmacokinetics of many drugs in children is different to that observed in adults, and it can change over time as the body matures. In children, many antiretrovirals are administered in mg/kg or mg/m2, but this does not guarantee adequate concentrations and therapeutic drug monitoring may be recommended to administer a dose which would provide adequate concentrations. In a review of clinical studies which analyse ART in children, 4 of the 23 studies adjusted the dose according to pharmacokinetic parameters. In these studies, the therapeutic efficacy was greater than that of other studies which used fixed doses136. In children, therapeutic drug monitoring has also proved useful to ascertain adherence.
Inhibitory quotient
When we use the Cmin referring to the theoretical EC50 of the sensitive virus or other similar methods for the therapeutic drug monitoring of PIs, we presuppose that the virus of the patient is completely sensitive to the drug we are monitoring. This may be the case in naïve patients102-104, but it is often not the case in patients who have presented one or more virological failures. Despite the methodological doubts they present, therapeutic drug monitoring studies which have used these parameters in patients with previous virological failure have not shown the strategy to be useful105-107. In patients who receive PIs, virological failures usually involve the emergence of mutations which reduce viral susceptibility to PIs, so that the drug concentration which is effective against sensitive viruses is not effective against viruses with reduced susceptibility. To solve this problem, the concept of inhibitory quotient (IQ) has appeared143.
The IQ is a parameter which relates the concentration of the drug to the susceptibility of the virus. It is a quotient whose numerator corresponds to exposure to the drug and whose denominator corresponds to resistance to the virus. Thus, the greater the concentration of the drug and the lower the resistance to the virus, the greater will be the IQ, whereas the lower the concentration and the greater the resistance, the lower will be the IQ. As a measure of exposure to the drug, any one of the pharmacokinetic parameters (AUC, Cmax or Cmin) can be used, although Cmin has traditionally been the most common. As a measure of resistance, the EC50 is used (IC50 corrected for plasma protein binding). The IQ tells us, therefore, the number of times that the Cmin of the drug is above the concentration necessary to inhibit 50% of viral replication and, in theory, would be a good parameter to evaluate whether a drug will be effective against a virus with specific susceptibility.
One of the main problems of using the IQ as a reference parameter for therapeutic drug monitoring is the need to phenotype the virus. The technique is very complex, expensive and can only be carried out in a few laboratories worldwide. Therefore, its application in clinical practice is impossible. To solve this problem, the so-called virtual inhibitory quotient (vIQ) has been developed, which, instead of using the real phenotype, uses the virtual phenotype. The virtual phenotype is an interpretation of the genotype which can be made thanks to a database with many genotypes and with the real phenotype corresponding to this genotype. Thus, in order to know the virtual phenotype, we need only know the genotype of the virus and apply a computer programme which will allow us to interpret it. The result is the number of times the susceptibility of the virus is reduced. To calculate the denominator of the vIQ, we must multiply the value of the virtual phenotype by the EC50 of the sensitive virus referred to in the literature.
The IQ50 corrected by the binding of the drug to plasma proteins intervenes in both the IQ and the vIQ. As mentioned above, this corrected IQ50 is not standardised and can be calculated in different ways with very different results. In order to avoid this problem, the normalized inhibitory quotient (nIQ) has been developed. The numerator is the vIQ and the denominator is the reference vIQ corresponding to the vIQ which, in the studies carried out to date, has proven useful in predicting the efficacy of the drug. The same corrected IC50 intervenes in both the numerator and denominator and can therefore be eliminated. Thus, the numerator is left with the product of the Cmin by the phenotype (number of times susceptibility is reduced) and the denominator with a different reference value for each drug, which corresponds to the product of the population Cmin and the phenotype below which the drug conserves its clinical efficacy. This reference value has been only been established for a few drugs.
Another inhibitory quotient has recently been defined, which uses the genotype as a measure of viral resistance, thus avoiding the need to have the real or virtual phenotype. This quotient is known as the genotypic inhibitory quotient gIQ, which is defined as the quotient of the Cmin and the number of mutations which reduce the susceptibility of the drug.
Studies to date144-161 have observed that either of the inhibitory quotients mentioned relates much better than the isolated Cmin to the therapeutic response to different PIs in patients with previous virological failure and reduced viral susceptibility. Undoubtedly, the inhibitory quotient will be very useful in this setting, although the wide variety of ways to calculate the inhibitory quotient and the requirements of some of them make it necessary to agree on a standard before applying it to normal clinical practice162-165.
Table 2 shows the different types of inhibitory quotient and their correlation with response to therapy.

Final comments and conclusions
1. Individual dosing according to plasma concentration is a therapeutic strategy has aroused interest in improving the efficacy of ART and in reducing its toxicity.
2. The immediate objective of therapeutic drug monitoring is the detection and correction of pharmacokinetic problems. Therapy does not only involve increasing or decreasing the dose of a drug, but it must also affect the underlying pharmacokinetic problem in each case (e.g. changing another drug which interacts with antiretrovirals, patient education on specific problems, etc.). We must not forget that therapeutic drug monitoring is not a strategy for treating the drug concentration but for treating the patient. For example, if the drug concentration is very high but the patient does not present toxicity, it would be difficult to find reasons to reduce the dose. Sometimes, therapeutic drug monitoring has been used as a complementary parameter to determine adherence to treatment.
3. The criteria a drug must fulfil to be suitable for therapeutic drug monitoring are:
a) Possibility of determining the drug concentration using accurate laboratory methods which are available in normal clinical practice.
b) Good relationship between plasma concentration, therapeutic efficacy and the appearance of adverse effects, in such a way that maintaining this concentration within specific margins can be beneficial.
c) Narrow therapeutic margin, with a relative facility for presenting sub-therapeutic or toxic concentration.
d) Wide interpatient variability and scarce intrapatient variability of the drug concentrations.
4. PIs and NNRTIs fulfil the suitability criteria for therapeutic drug monitoring. NRTIs do not and cannot currently be considered suitable for therapeutic drug monitoring.
5. Limitations to therapeutic drug monitoring include:
a) Confusion between pharmacokinetic problems which lead to a low drug concentration and poor adherence.
b) Inadequately defined parameters for measuring exposure to the drug and its efficacious concentration. Different pharmacokinetic parameters have been used to measure exposure to the drug, and a single sample, usually Cmin, is recommended in clinical practice. It may be difficult to know exactly when to take the sample with respect to taking the medicine (this is more important for PIs than for NNRTIs). The concentration determined must be a concentration which is considered therapeutic. This can be obtained by mathematical calculations using theoretical concentration-time curves, but the IQ50 corrected by binding to plasma proteins, which is found in the literature, is usually taken as a reference. This correction can be made in different ways, with different results. Moreover, the free fraction of the drug can vary in certain circumstances. In the case of nelfinavir, the presence of an active metabolite in considerable quantities can make it difficult to interpret the concentration of the drug. In any case, the necessary concentration must be interpreted on an individual basis, taking into account the fact that patients with partially resistant viruses will need higher concentrations and that the efficacy of treatment will depend on all the drugs involved and the synergy between them.
6. Very few randomized clinical trials have been carried out to evaluate the use of therapeutic drug monitoring, although those involving naive patients have shown that therapeutic drug monitoring improves the efficacy of treatment. The studies carried out in patients with therapeutic failure have not observed differences in efficacy, although they are affected by important problems of approach and methodology which make it impossible to interpret the results. One of the most important problems is the choice of efficacious reference concentrations, which may not be useful for partially resistant viruses.
7. The concept of inhibitory quotient includes both pharmacokinetic data and viral resistance data. In this way we can evaluate whether the plasma concentration of the drug is the appropriate concentration for a virus with a specific susceptibility. The inhibitory quotient is related to the efficacy of treatment in patients with virus who present reduced susceptibility and may be useful for therapeutic drug monitoring. Nevertheless, methodological standardization and a clinical validation are necessary before applying it in normal clinical practice.
8. Therapeutic drug monitoring will be particularly useful when the risk of sub-therapeutic or toxic concentrations is very high. Therefore, it is recommended in patients with any of the following characteristics:
a) Suspected clinically important pharmacokinetic interactions.
b) Suspected intestinal malabsorption.
c) Appearance of adverse effects which can improve with a reduced plasma concentration of the drug, especially in low-weight patients, women and in patients with cirrhosis of the liver.
d) Virological failure with no evident cause.
e) Pregnant women and children, whose pharmacokinetics can suffer modifications.
f) Patients who start ART with drugs which have relatively low concentrations, such as non-boosted PIs or once-daily treatments with some boosted PIs.
9. Boosting of PIs with ritonavir considerably increases their plasma concentration, although this does not invalidate their usefulness in therapeutic drug monitoring, given that a high pharmacokinetic variability may persist and viruses with reduced susceptibility may need higher concentrations of PI.
10. It is important that any therapeutic drug monitoring program contain measures to monitor and improve adherence, thus making it easier to interpret concentrations and provide a suitable response to therapy.
11. In summary, there is a clear basis for thinking that therapeutic drug monitoring may be a useful tool for optimising ART in certain circumstances. Before recommending its widespread application as a routine method in normal clinical practice, it is necessary to standardise the parameters which must be used and carry out methodologically correct studies with a high number of patients to profile the role of therapeutic drug monitoring in different clinical situations. It may also be necessary to develop education programs for physicians, pharmacists, healthcare personnel and patients, so that the information obtained from monitoring can be used properly.
Acknowledgements
We are grateful to Dr. Rosa M.ª López Galera for her collaboration in the preparation of this manuscript.

