




Yersinia enterocolitica is a heterogeneous group of strains, which are classified into 6 biogroups, and into more than 57 O serogroups. However, the human pathogenic strains most frequently isolated worldwide belong to serogroups O:3, O:5,27, O:8 and O:9. The major route of Y. enterocolitica infection is through contaminated foods or water. The primary pathogenic event is colonization of the intestinal tract where most of the pathologic effects and clinical manifestations occur. Temperature and calcium concentration regulate expression of virulence factors that guide the invading yersiniae and allow them to survive and disseminate. Gastrointestinal infections are usually self-limiting and do not merit antimicrobial therapy. Nonetheless, fluoroquinolones or third generation cephalosporins, the best therapeutic options, are warranted to treat enterocolitis in compromised hosts and in patients with septicemia or invasive infection, in which the mortality can be as high as 50%. A review of the pathogenesis, virulence and antimicrobial resistance is carried out.
Y. enterocolitica es un grupo heterogéneo de cepas clasificadas en 6 biogrupos y en más de 57 serogrupos O. Las cepas patógenas humanas más frecuentemente aisladas pertenecen a los serogrupos O:3, O:5,27, O:8 y O:9. La transmisión de la infección es principalmente a través de alimentos o agua contaminados. La etapa esencial de la patogénesis es la colonización del tracto intestinal, donde ocurren la mayoría de los efectos patológicos y manifestaciones clínicas. La temperatura y la concentración de calcio regulan la expresión de los factores de virulencia que guían al patógeno durante la invasión, supervivencia y diseminación. Normalmente las infecciones gastrointestinales son autolimitadas y no necesitan tratamiento antimicrobiano. Las fluoroquinolonas y cefalosporinas de tercera generación son los tratamientos más eficaces en casos de enterocolitis en immunodeprimidos, septicemia o infección invasiva, situaciones en las que la mortalidad puede alcanzar el 50%. Se presenta una revisión de la patogénesis, virulencia y resistencia antimicrobiana.
Bacteria of the genus Yersinia cause diseases ranging from enteritis to bubonic plague (Black Death). The initial characterization of this genus was performed in 1894 in Hong Kong. There, Alexandre Emile John Yersin together with Shibasaburo Kitasato identified Yersinia pestis (formerly known as Pasteurella pestis) as the causal agent of the bubonic plague.1 However, the first recognized description of 5 human isolates belonging to Yersinia enterocolitica was made later, in 1939, by Schleifstein and Coleman in the United States.2 Nonetheless, McIver and Pike had already isolated one of these clinical strains in 1934, although they had erroneously identified it under the name Flavobacterium pseudomallei.3
Members of the genus Yersinia are non-spore forming, Gram-negative or Gram-variable, rod-shaped or coccoid cells of 0.5–0.8μm in width and 1–3μm in length. All species, with the exception of Y. pestis, are motile at 22–30°C but not at 37°C. Motile cells are peritrichously flagellated. Yersiniae grow under aerobic and anaerobic culture conditions between 0°C and 45°C, being optimum at 25–28°C, on non-selective and certain selective media.4
At present, the genus Yersinia includes 11 established species: Y. pestis, Y. pseudotuberculosis, Y. enterocolitica, Y. frederiksenii, Y. intermedia, Y. kristensenii, Y. bercovieri, Y. mollaretii, Y. rohdei, Y. aldovae and Y. ruckeri. Among them only Y. pestis, Y. pseudotuberculosis and certain strains of Y. enterocolitica are of pathogenic importance for humans and certain warm-blooded animals, whereas the other species are of environmental origin and may, at best, act as opportunists. However, Yersinia strains can be isolated from clinical materials and therefore have to be identified at the species level.4
Y. enterocolitica is a heterogeneous group of strains, which are traditionally classified by biotyping into 6 biogroups on the basis of phenotypic characteristics, and by serotyping into more than 57 O serogroups, on the basis of their O (lipopolysaccharide or LPS) surface antigen. Five of the 6 biogroups (1B and 2–5) are regarded as pathogens. However, only a few of these serogroups have been associated with disease in either humans or animals. Strains that belong to serogroups O:3 (biogroup 4), O:5,27 (biogroups 2 and 3), O:8 (biogroup 1B) and O:9 (biogroup 2) are most frequently isolated worldwide from human samples.5 However, the most important Y. enterocolitica serogroup in many European countries is serogroup O:3 followed by O:9, whereas the serogroup O:8 is mainly detected in the United States.1
Y. enterocolitica is widespread in nature, occurring in reservoirs ranging from the intestinal tracts of numerous mammals, avian species, cold-blooded species, and even from terrestrial and aquatic niches. Most environmental isolates are avirulent; however, isolates recovered from porcine sources contain human pathogenic serogroups. In addition, other studies have suggested that dogs, sheep, wild rodents and environmental water may also be a reservoir of pathogenic Y. enterocolitica strains. Human pathogenic strains are usually confined to the intestinal tract and lead to enteritis/diarrhea.6
Clinical identificationClinical identification of Y. enterocolitica strains is achieved after growth of stool samples on MacConkey plates, as well as on CIN Agar (Cefsulodin, Irgasan, Novobiocin), also known as Yersinia Selective Agar. Plates are incubated for 24h at 37°C and identification of colonies is performed according to macroscopic characteristics. Y. enterocolitica has slower lactose fermentation and slower growth in comparison with other enteric bacterial species normally present in fecal samples. Therefore, the pinpoint and colorless colonies grown on MacConkey plates make identification a difficult task against the background of the more rapidly growing bacteria.1 Consequently, the use of selective media such as the CIN Agar, which is a differential medium that inhibits growth of normal enteric organisms, provides improved direct recovery of this pathogen from feces. The characteristic translucent and sharp-bordered colonies, showing a deep-red center due to mannitol fermentation, are usually referred to as bull's eyes colonies and allow easy identification.7
In addition, there are specific biochemical properties which corroborate its identification. The most significant traits are the following: Y. enterocolitica strains are motile at 25°C but not at 37°C, produce urease, lack oxidase activity and do not produce either gas or hydrogen sulphide on Kligler's Iron Agar.1
In order to distinguish between pathogenic and non-pathogenic strains, further approaches can be undertaken. Serogroup analysis can be performed using specific antisera for typing pathogenic strains. According to the geographical distribution of the serogroups, this methodology is particularly important in Europe or Japan to detect serogroups O:3 and O:9. On the other hand, this serological diagnostic method has been minimally used in the United States to detect serogroup O:8 because of the absence of sufficient guidelines for interpretation of agglutinin titers.1,8
When antisera are not available, assessment of virulence traits is an alternative methodology and generally identifies expression of plasmid-encoded virulence factors by means of simple phenotypic tests. Pathogenic strains are usually positive for autoagglutination at 37°C, calcium dependence for growth at 37°C, Congo red binding, and resistance to the bactericidal effects of serum.9–11 However, absence of pyrazinamidase production and lack of salicin fermentation and esculin hydrolysis are traits of pathogenic yersiniae independently associated with the presence of the virulence plasmid.12
Nowadays, MALDI-TOF mass spectrometry analysis has been incorporated as a straightforward, rapid, accurate and inexpensive tool to identify bacteria according to specific protein profiles. Therefore, it is currently displacing the former biochemical and phenotypic characterization in an increasing number of clinical microbiology laboratories.13,14
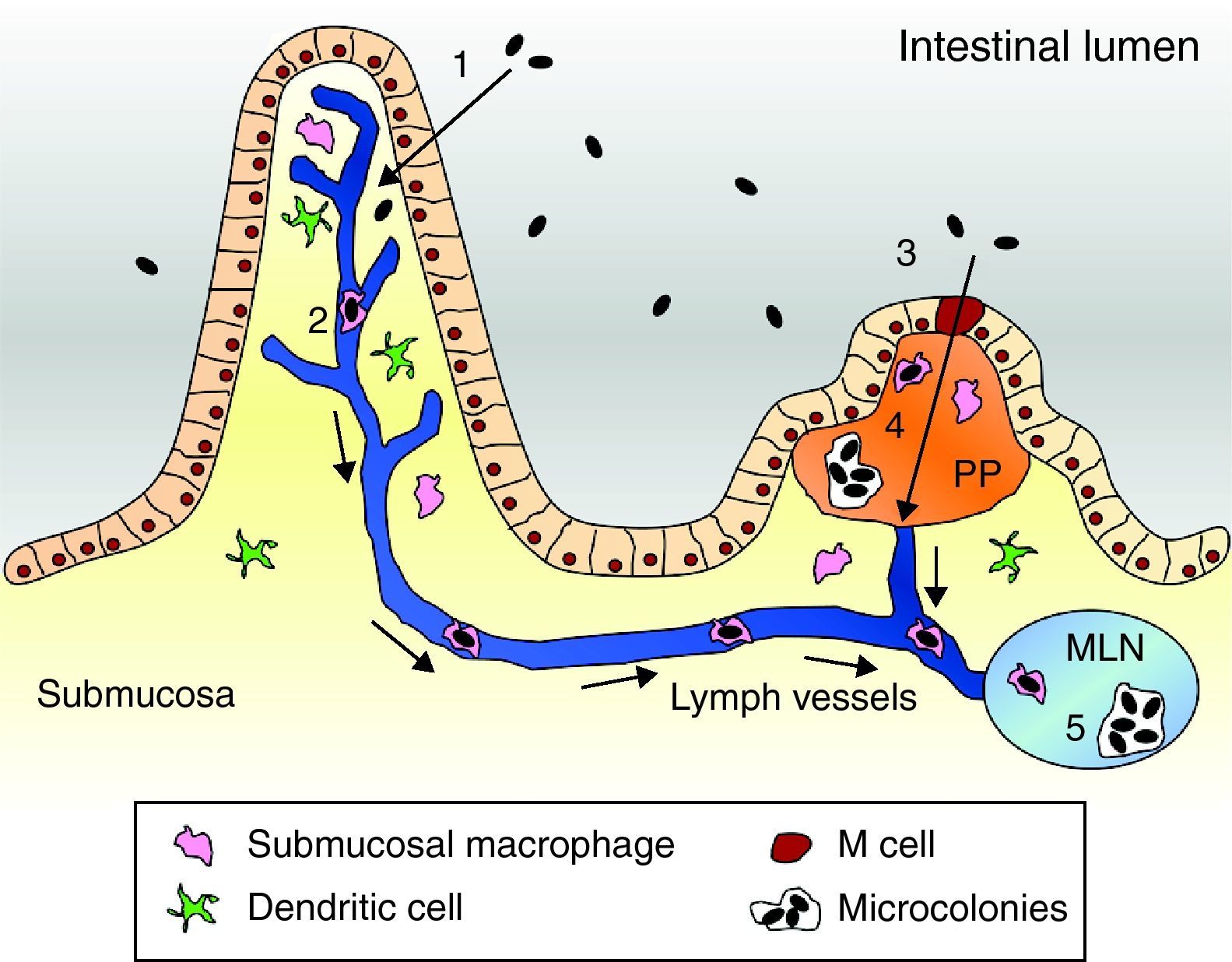
Pathogenesis modelThe usual route of acquisition of this pathogen is through contaminated foods or water.15,16 The primary event of Y. enterocolitica pathogenesis is colonization of the intestinal tract, particularly the distal small intestine (terminal ileum) and proximal colon. Accordingly, most of the pathologic effects and, hence, clinical manifestations occur at this location.1 There, virulent yersiniae must traverse the intestinal lumen, attach to and penetrate the mucus barrier overlying the mucosal epithelial cells, and eventually adhere to intestinal cells. Bacteria have shown to preferentially bind to and penetrate M cells of Peyer's patches (PP). Once internalized, the bacteria are transported across the epithelial barrier and expelled from the basolateral side of the M cell.1,17,18 There, evidence suggests that in the earliest steps of infection, phagocytes internalize the bacteria. Internalized bacteria reportedly replicate in native murine macrophages and hence they are assumed to be transported within migrating phagocytes to mesenteric lymph nodes (MLN), causing an inflammatory response that triggers abdominal pain.19,20 Furthermore, phagocytes that take up bacteria can disseminate via the bloodstream to the liver and spleen. Once located in PPs, MLNs, the spleen or liver, Y. enterocolitica replicate in an extracellular form within micro-abscesses.21,22 Within these lesions the bacteria form microcolonies and appear to be resistant to phagocytosis by macrophages and neutrophils23 (Fig. 1).
 Yersinia cells traverse the intestinal epithelium via epithelial cells to the submucosa. (2) Submucosal macrophages phagocytose the pathogen and enter into the lymphatic system thereby reaching the MLN. (3) Alternatively, bacteria can be engulfed by M cells. (4) Once in the PP Yersinia forms microcolonies and starts replication. (5) Eventually, bacterial cells are located in the MLN and can equally form microcolonies to allow replication.")
Pathogenesis model of Yersinia enterocolitica. (1) Yersinia cells traverse the intestinal epithelium via epithelial cells to the submucosa. (2) Submucosal macrophages phagocytose the pathogen and enter into the lymphatic system thereby reaching the MLN. (3) Alternatively, bacteria can be engulfed by M cells. (4) Once in the PP Yersinia forms microcolonies and starts replication. (5) Eventually, bacterial cells are located in the MLN and can equally form microcolonies to allow replication.
The virulence factors characterized in Y. enterocolitica are located within the chromosome and also on a 70kb virulence plasmid designated pYV which is only detected in virulent strains. These virulence properties, particularly the plasmid-encoded genes, guide the invading Yersinia pathogen and enable bacteria to survive inside the human host.1,24,25
Adhesion/invasion factorsFor the initial step of invasion of the intestinal mucosa, three proteins have been shown to take over the process and hence allow intimate attachment to the epithelial cells. The invasin per se, Inv, is the major determinant that plays a vital role by promoting entry of bacteria into epithelial cells.26,27 However, experiments suggest that there must be an alternative factor leading to occasional colonization of PP in a less efficient invasion pathway.27 YadA, a pYV plasmid-encoded protein, has been described as the major adhesion for attachment, being essential for induction of disease (e.g., inflammation and necrosis in the liver). It is a multifaceted protein that mediates adherence to epithelial cells, phagocytes and extracellular matrix components, and protects the bacterium being killed by neutrophils. Moreover, it is involved in autoagglutination, a phenomenon occurring after growth in tissue culture medium at 37°C.28,29 After invasion, YadA predominates as adhesion in infected tissue and is required for persistence, survival and replication in PP.30 In addition, a certain invasion ability has been attributed to this protein, particularly detectable only after inv inactivation.30,31 The third protein, Ail, is highly correlated with virulence, since it has only been detected among pathogenic Yersinia strains, in comparison with the inv gene which is found in all Yersinia isolates. Ail is involved in adhesion to and invasion of specific tissue culture cells, as well as in survival against the bactericidal effects of serum.26,32
FlagellaBefore Y. enterocolitica establishes intimate contact with the intestinal epithelium, it has been reported that flagella and, hence, motility play an important role in initiating host cell invasion. Inactivation of the flagellar regulatory genes, either flhDC (the master regulatory component) or fliA, all required for the expression of motility, has been associated with a decrease in invasion comparable to that of strains in which inv has been inactivated. In agreement with these results, if bacteria are then brought into contact with the in vitro monolayer of eukaryotic cells by centrifugation, no difference can be detected between the flagellar mutants and the wild-type strain concerning invasion (despite impaired invasion still being detected for the inv mutant).33
Lipopolysaccharide (LPS)The LPS is the major component of the outer membrane of Gram-negative bacteria. It is composed of: (i) lipid A, anchored into the membrane and associated with toxicity; (ii) the core, including both inner and outer moieties (mainly sugars); and (iii) the O antigen or O polysaccharide chain, the external component associated with antigenic properties. Nonetheless, several studies have also reported that the O antigen is involved in the colonization and invasion processes since O antigen-deficient strains (serotypes O:3 and O:8) are attenuated after oral infection of mice. These mutants display an inefficient colonization of PP in comparison to wild-type strains and do not multiply in the spleen, liver and MLN. Interestingly, the authors show that YadA function is impaired in the absence of the O antigen, Ail is not expressed, and Inv is down-regulated. Thus, it seems that the O antigen is required for the proper expression or functionality of other outer membrane virulence factors justifying the diminished host cell internalization.34,35
Ysc T3SSOn the other hand, the pYV plasmid encodes the Yop virulon, which is the core of Yersinia pathogenicity machinery.25 The Yop virulon comprises the Yop effectors, which are delivered to the extracellular milieu, the plasma membrane, or into the host cell cytosol, and the corresponding secretion machinery called Ysc T3SS, which includes the injectisome, the apparatus that spans both bacterial membranes, and the translocators YopB, YopD and LcrV.36,37 The injectisome consists of a large number of Ysc proteins, encoded on genes that are clustered in three large neighboring operons called virA, virB and virC that constitute the needle complex and the basal body. Secretion of some Yops requires the presence of small cytosolic chaperones, which bind to a specific partner Yop, and belong to a family called the Syc proteins: SycE (for YopE), SycH (for YopH), SycT (for YopT), SycN (for YopN) and SycD (for YopB and YopD). In the absence of these chaperones Yop secretion is severely reduced, if not abolished.38
The Ysc T3SS counteracts several key innate defense mechanisms of phagocytes and down-regulates inflammation. Four of the six effectors identified so far (YopH,39 YopE,40 YopT41 and YopO42) inhibit cytoskeleton dynamics and contribute to the strong resistance of pathogenic Yersinia to phagocytosis by macrophages and neutrophils.43 In general, when one of these four Yops is lacking, bacteria are more efficiently phagocytosed indicating that there is no redundancy between the Yops, but rather synergy. YopH antagonizes several signaling pathways important for innate and adaptive immunity. This protein reduces bacterial internalization and killing by neutrophils or macrophages by counteracting responses associated with phagocytosis, such as the oxidative burst, and preventing the production of proinflammatory cytokines.38,43
The three other Yop effectors act on signaling pathways that control gene expression, as well as the dynamics of the cytoskeleton. YopE causes disruption of the actin cytoskeleton and thus rounding and detachment of infected cells in culture, a phenomenon traditionally called cytotoxicity.40 In addition to counteracting phagocytosis, YopE inhibits proinflammatory cytokine production in infected epithelial cells together with other Yops. A yopE mutant, as well as a yopH mutant, is more efficiently internalized and more rapidly eliminated from the liver and spleen. YopT shows similar effects to those of YopE in cultured cells: disruption of stress fibers, cell rounding and inhibition of phagocytosis.41 However, in a mouse model, a yopT mutant is as virulent as its parental strain and disseminates to the spleen and liver, killing the mice with similar kinetics as those of the wild-type. These findings suggest that YopE and YopT share overlapping virulence functions and, therefore, some level of functional redundancy. Activated by actin binding, YopO triggers cytotoxicity of cultured cells, albeit only when the protein is introduced at high levels in the absence of other effector proteins. Furthermore, it also contributes to the anti-phagocytic activity, despite the report of controversial results concerning virulence in in vivo models.20,38,43
Yop effectors, YopP44 and YopM,45 also promote the intracellular survival of Yersinia by counteracting the production of proinflammatory cytokines and thus blocking the ability of the cell to respond to infection. YopP suppresses production of TNF-alpha by macrophages and IL-8 by epithelial and endothelial cells. It also induces apoptosis of macrophages and presumably reduces neutrophil recruitment to the site of infection.44 YopP is important for virulence when administered orally in in vivo models, since the reduced virulence upon yopP inactivation is not associated with a significant reduction in systemic spread. YopM function has not yet been completely defined. It has been shown to migrate to the nucleus of target cells, despite controversy about its possible role in affecting gene transcription. In addition, it has been shown to interact with cytoplasmic kinases and may be involved in reducing the levels of IL-10 and IL-18. Nonetheless, YopM is required for virulence since a yopM mutant is unable to establish systemic infection following oral challenge of mice. Furthermore, YopH also contributes to the down-regulation of inflammatory response by inhibiting the recruitment of other macrophages to the sites of infection.20,38
Ysa T3SSWhile the Ysc T3SS is important for the virulence of Yersinia during systemic stages of infection, it is now clear that some isolates utilize additional T3SSs. The Y. enterocolitica biotype 1B carries the Ysa pathogenicity island (Ysa-PI) encoding a new T3SS.46 The Ysa T3SS plays an important role in the colonization of gastrointestinal tissues during the earliest stages of infection and facilitates the overcoming of immune barriers presented by the host at this location.47
The ysa genes encode this recently described T3SS apparatus, which is closely related to the T3SS-1 encoded within the Salmonella enterica SPI-1 and the Mxi-Spa T3SS of Shigella. The translocated and effector proteins are termed Ysp and according to this homology and nomenclature, the YspB, YspC and YspD proteins encoded within the sycByspBCDA operon constitute the translocon complex, whereas SycB is described as a chaperone.48,49
The first effectors characterized to be exported by the Ysa T3SS were termed YspG, YspH and YspJ. Nonetheless, these proteins turned out to be indeed the plasmid-encoded determinants YopN, YopP and YopE, respectively.50 Since then, up to 16 effectors have been identified (YspA, YspE, YspF, YspI, YspK, YspL, YspM, YspN, YspP and YspY, also including the Yops and the translocon members). The corresponding genes are strikingly dispersed throughout the genome (within the Ysa-PI, on the pYV plasmid and dispersed on the chromosome).51,52 These proteins have been shown to be necessary for full virulence, since individual ysp mutants are deficient in the colonization of gastrointestinal tissues in in vivo models.52 However, their homology to other T3SS and function is still under study to identify their cellular targets and, hence, their role in the pathogenesis.
Among the ysa genes, ysaE encodes a protein that belongs to the AraC family of transcriptional regulators which has been reported to act in concert with the chaperone SycB to activate the operon sycByspBCDA as well as other Ysp effectors.53 This phenomenon is similar to that reported in Salmonella, in which InvF (the AraC-like regulator) acts together with the chaperone SicA to activate downstream genes.54 Moreover, a peculiar “two” component regulatory system near the ysa genes has been reported to be required for expression of the ysaE gene. The two adjacent genes, ysrS and ysrR, encode the sensor kinase and the response regulator, respectively, thought to represent a key regulatory component for the secretion of the Ysa apparatus.53 In general terms, these systems are composed of two proteins in which the sensor kinase is autophosphorylated at a conserved histidine residue (H) and the phosphoryl group is then transferred to a specific aspartic residue (D) within the response regulator. In particular situations, these systems are termed multi-component phosphorelay systems, since the sensor kinase is, in fact, a hybrid protein that includes additional sites for phosphorylation (H1, D1 and H2) and, hence, transference of the phosphoryl group.55 In this case, however, the histidine phosphotransferase domain, where the H2 site is located, is not found in YsrS, but instead it has been characterized within a different protein termed YsrT (encoded by a gene adjacent to ysrS). Since introduction of an alanine substitution at H2 (within YsrT) leads to loss of activation of ysaE expression, these results corroborate that the three proteins are members of this phosphorelay system.56
Enterotoxin YstY. enterocolitica is also known to produce a heat-stable chromosomally encoded enterotoxin, known as Yst. Thus far, several antigenically related variants have been characterized. However, the role of this toxin in diarrheal disease remains largely controversial: (i) Yst cannot be detected in diarrheal stool samples in experimental animal models after infection with Y. enterocolitica; (ii) some strains carry the yst gene, but fail to produce the enterotoxin, suggesting the presence of silent genes; and (iii) the proportions of the enterotoxigenic bacteria are similar among clinical and non-clinical isolates. Nonetheless, non-invasive biotype 1A strains causing diarrhea frequently carry a variant of the yst gene, which could be the only virulence factor accounting for this diarrheal illness.1,57
High-Pathogenicity Island (HPI)A further set of virulence genes is that encoded within another pathogenicity island, termed High-Pathogenicity Island (HPI). Similarly to the Ysa-PI location, this chromosomal region is only present in Y. enterocolitica biotype 1B. Most of the genes located on this island are involved in the biosynthesis, transport and regulation of the siderophore yersiniabactin. Thus, the HPI may be regarded as an iron-capture island.58 The locus involved is composed of 11 genes organized into four operons (fyuA, irp2, ybtA and ybtP) which can be divided into three functional groups: yersiniabactin biosynthesis, transport into the bacterial cell (outer membrane receptor and transporters), and regulation.59 The most representative proteins include: FyuA, the outer membrane receptor for yersiniabactin60; and the inner-membrane permeases YbtP and YbtQ required for the translocation of iron into the bacterial cytosol.61,62 On the other hand, YbtA is the AraC transcriptional regulator which activates expression from the three other promoters (fyuA, irp2 and ybtP), although it represses its own expression.63,64 Alternatively, there is some evidence that yersiniabactin itself may up-regulate its own expression and that of fyuA. In addition, all four promoter regions possess a Fur-binding site and are negatively regulated by this repressor in the presence of iron.59
The myf operonLastly, the chromosomal locus called myf has been reported to encode several genes, such as myfA, myfB and myfC, which constitute a fibrillar structure, which closely resembles CS3 fimbriae of ETEC. MyfA represents the major subunit. The assembly machine includes MyfB, the putative chaperone, and MyfC, the membrane usher protein. Experiments performed with Y. pseudotuberculosis attribute a role in thermoinducible binding and haemagglutination to the psa locus (the myf homolog). However, in Y. enterocolitica the myf operon is not able to mediate haemagglutination and further experiments regarding its adhesive function and its possible role in pathogenesis are yet to be performed.65,66
Regulation of virulenceAll pathogenic Y. enterocolitica strains, both low and high-pathogenicity isolates, show a thermally responsive adaptation process which aids the transition from the environment to the adverse conditions inside of the human host. The rapidity of this process in surviving yersiniae cells determines the clinical outcome, as well as the incubation period.1 Thus, regulation of virulence genes plays a key role in the successful accommodation for the increase in temperature to 37°C when infecting eukaryotic tissues. In consequence, several plasmid-encoded factors are expressed at 37°C but not at 26°C. Furthermore, the presence or absence of calcium in the intracellular environment or culture media is also an important regulatory mechanism of virulence. Nonetheless, these two environmental signals influence two different regulatory networks.67,68
Inv, which is expressed at environmental temperatures, is used to establish initial colonization, and, as acclimatization to mammalian host temperatures ensues, Ail and plasmid-encoded determinants, such as YadA, the Yops and the virA, virB and virC operons, are gradually expressed to contribute in the establishment of the infection as well as in combating host defense mechanisms.32,67,69,70 Therefore, once bacteria reach a temperature of 37°C, the Ysc injectisomes are assembled and a stock of intracellular Yops is synthesized. However, secretion of effectors only starts when bacteria establish close contact with target cells and docks at eukaryotic cell surfaces by means of Inv and YadA activity. In the case of phagocytes, the adhesions are dispensable because phagocytic receptors provide the necessary intimate contact. Then, the secretion channel opens, the translocator proteins are delivered into the target cell plasma membrane and eventually the effector Yops are exported.38
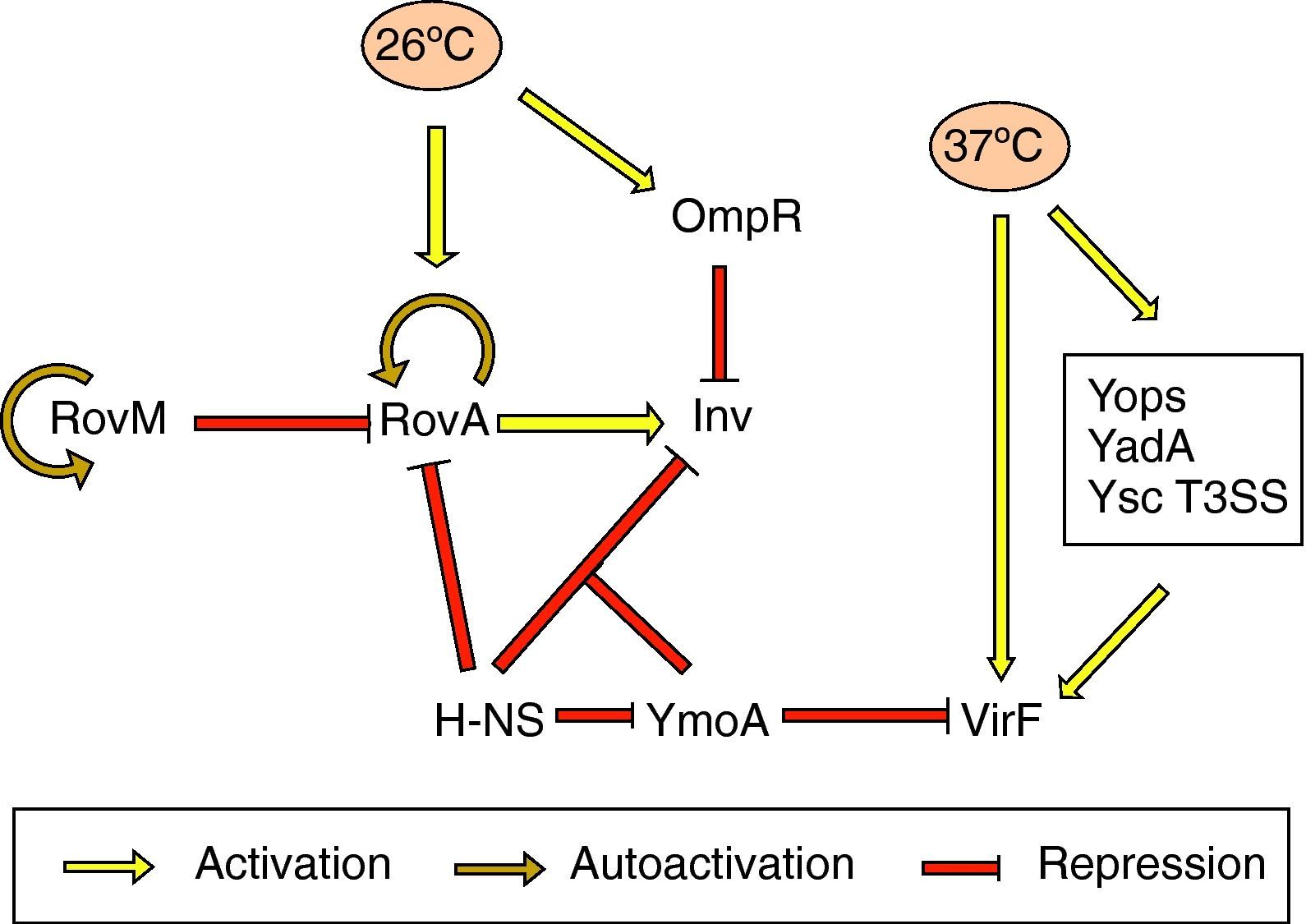
Effect of temperatureInvInv expression is regulated in response to pH, growth phase and temperature. There is in vitro maximum inv expression at 26°C, pH 8.0, or 37°C, pH 5.5, in early stationary phase. Thus, Inv is maximally expressed at the environmental temperatures 25–28°C.69 Thermoregulation depends on the levels of RovA, a MarR-type regulator that activates inv expression in vivo and in vitro.71 In Y. pseudotuberculosis RovA has been shown to activate itself in response to environmental signals, such as moderate temperature (20–28°C), stationary phase growth and nutrient rich growth medium by binding to its own promoter.72 In addition, rovA expression is under the control of several regulators. RovM, a LysR-like transcriptional repressor which has its homolog in Y. enterocolitica, interacts with the rovA promoter and negatively regulates rovA transcription. rovM itself is under positive autoregulatory control and is significantly induced during growth in minimal media. However, it has been deduced that temperature-dependent rovA expression does not occur through RovM.73 Furthermore, rovA transcription is subject to silencing by the H-NS protein. RovM and H-NS bind simultaneously to different regions of the rovA promoter and thus cooperate for efficient silencing of the rovA gene in Y. pseudotuberculosis. On the other hand, RovA and H-NS compete in binding to the same region in such a way that RovA acts as a derepressor by displacing H-NS. A similar competition between both proteins occurs on the inv promoter. As regards inv expression, the global regulator YmoA has been shown to interact with H-NS and form a repressor complex, with H-NS providing the binding specificity. Moreover, H-NS has been reported to repress ymoA transcription.74 The levels of H-NS and YmoA are similar between 26°C and 37°C, whereas the levels of RovA are only high at 26°C. Thus, Inv expression is governed by the levels of RovA within the cell and can only be derepressed at 26°C (Fig. 2).73,75 More recently, OmpR, the response regulator of the EnvZ/OmpR two-component system, has been reported to negatively regulate inv expression at 26°C. However, at the repressor temperature of 37°C, the OmpR protein plays no role in inv regulation.76
The yop stimulon
The pYV plasmid-encoded genes that are expressed at 37°C constitute the yop stimulon, and among them there is a subset of genes, e.g., all the yops, yadA and members of the T3SS apparatus that are regulated by VirF, and hence represent the VirF regulon.24 VirF is a transcriptional activator that belongs to the AraC family of regulators and is itself strongly thermoregulated and highly expressed at 37°C.70 Thus, expression of the yop stimulon is first controlled by temperature, and expression of some of its genes is reinforced by the action of VirF, in which synthesis is also temperature controlled. Another important protein involved in thermoregulation is YmoA. In the absence of this protein there is increased transcription of virF at 28°C and hence strong expression of the Yops and YadA. Thus, YmoA acts as a repressor at low temperatures (Fig. 2).77
Nonetheless, expression and/or secretion of other general and biotype-specific virulence determinants, such as the flagella and the Ysp effectors, respectively, also fit in this regulatory model mainly driven by temperature. It has been shown that secretion of the Ysp effector proteins only occurs when cells are incubated at 26°C.53 Similarly, motility is only displayed at 30°C or below, due to the rapid arrest of flagellin transcription when bacteria are incubated at 37°C.78 Moreover, in addition to negatively regulating inv expression, OmpR exerts a positive effect on flhDC expression by direct binding to the flagellar promoter (the OmpR function, however, is not related to temperature).79 Thus, according to their role in the pathogenesis process, the order in which the virulence determinants are expressed is important. Coincident with the timing of Yop expression at 37°C inside the host cell, bacteria are no longer motile and stop Ysp secretion revealing a model in which invasion and motility are inversely regulated.
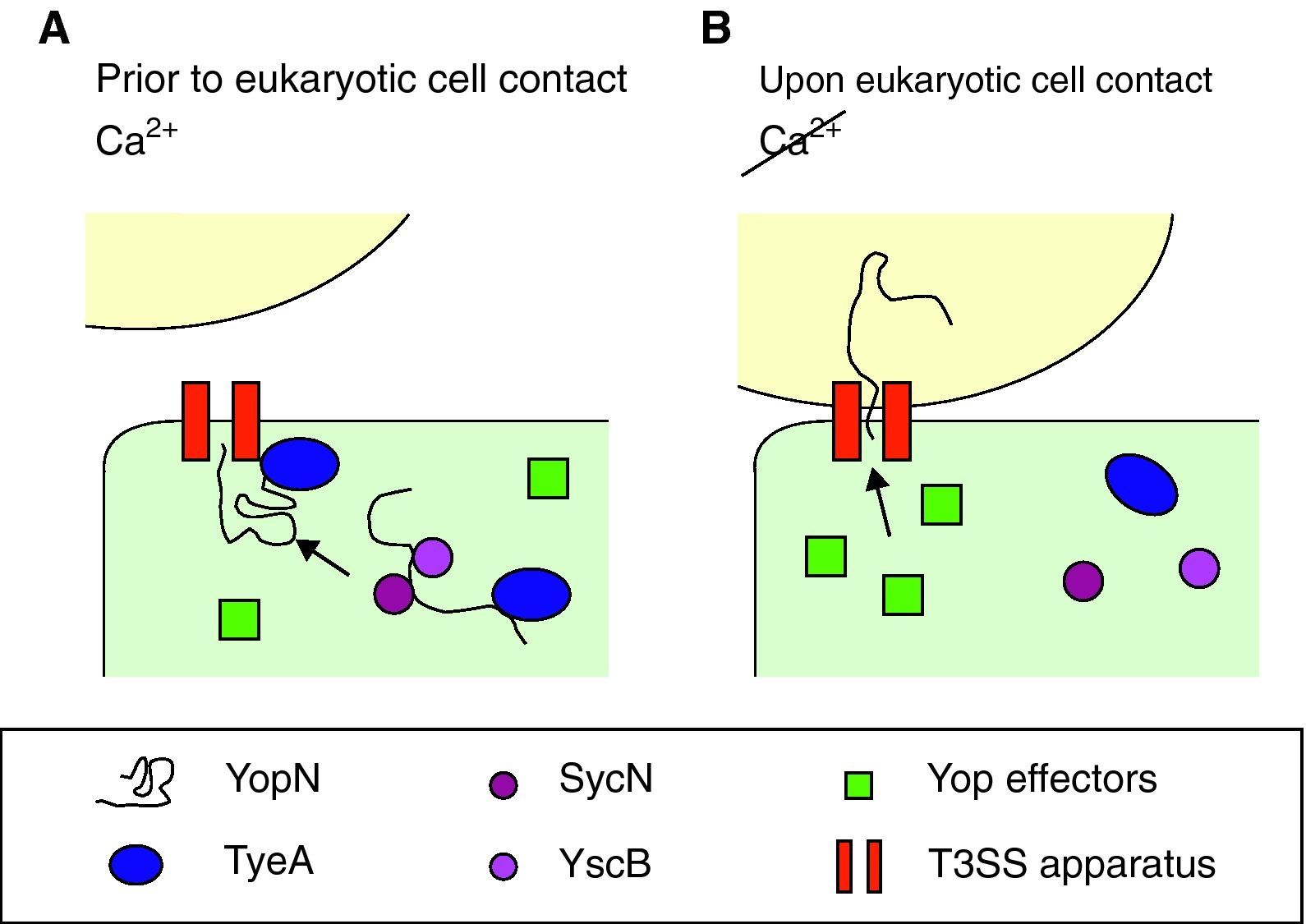
Effect of calciumYop secretion and expressionDespite production of Yops occurring at 37°C, they are only secreted in vitro in the absence of calcium, which correlates with growth arrest, a phenomenon long known as calcium dependency. However, in vivo secretion only occurs upon close contact with target cells by a mechanism involving calcium. Therefore, the presence of calcium ions blocks not only the secretion of Yops, but also their further synthesis. Bacteria take up yop transcription again only after contacting with the target cell has been established. As for the mechanism beyond calcium regulation, it has been suggested that calcium stops secretion whereas a feedback inhibition mechanism blocks transcription of the yop genes when secretion is compromised.24 Opening of the secretion channel relies on YopN transport, which plays a central role in the regulatory mechanism for the activity of the type III pathway, although the exact process is unknown. The model proposed by Ferracci et al. in Y. pestis suggests that the SycN/YscB chaperones target the YopN/TyeA intracellular complex to the T3SS machinery.80 There, in the presence of calcium and prior to contact with eukaryotic cells, the YopN/TyeA complex acts as a plug by blocking the Ysc secretion channel and preventing YopN transport (Fig. 3A). Under induction conditions, the YopN interactions are disrupted and hence YopN can be initiated into the type III pathway and open the secretion pathway to the effectors (Fig. 3B).80,81 Alternatively, YscM1 and YscM2 are two secreted negative regulators which are involved in the feedback inhibition mechanism. The absence of any of these proteins leads to increased Yop synthesis, even in the presence of calcium, whereas when YscM1 is overproduced, yop transcription is abolished.82
 Yersinia has acclimated to mammalian host temperatures. The Ysc injectisome is installed and a stock of Yop proteins is synthesized. Nonetheless, the SycN/YscB chaperone complex has targeted the YopN/TyeA complex to the Ysc apparatus, hence blocking the effector transport. (B) Upon contact with the eukaryotic cell conformational changes allow the release of the YopN/TyeA complex from the Ysc apparatus so that the YopN protein can be transported to the eukaryotic cytoplasm. Secretion of the Yop effectors then follows.")
Optimal parameters and mechanism to start injection of the Ysc T3SS effectors. (A) Yersinia has acclimated to mammalian host temperatures. The Ysc injectisome is installed and a stock of Yop proteins is synthesized. Nonetheless, the SycN/YscB chaperone complex has targeted the YopN/TyeA complex to the Ysc apparatus, hence blocking the effector transport. (B) Upon contact with the eukaryotic cell conformational changes allow the release of the YopN/TyeA complex from the Ysc apparatus so that the YopN protein can be transported to the eukaryotic cytoplasm. Secretion of the Yop effectors then follows.
Among the different biotypes and throughout the gastrointestinal tract, Y. enterocolitica biotype 1B (mainly associated in the clinical setting with serotype O:8) is known as highly pathogenic and usually produces the most catastrophic events. However, serogroups O:3 and O:9 (belonging to biotypes 4 and 2, respectively), which are the most common causes of yersiniae infections worldwide, are less destructive.1 Since iron appears to play a crucial role in the pathogenesis of Yersinia, one of the major differences between low and high-pathogenicity strains lies in their ability to capture iron molecules. The presence of particular virulence factors, such as the HPI genes in addition to the Ysa T3SS, correlates with the level of pathogenicity. Both sets of genes are only detected in Y. enterocolitica biotype 1B. Accordingly, the presence of the yersiniabactin, the high-affinity iron-chelating system encoded within the HPI, allows the bacteria to multiply in the host and to cause systemic infections, whereas in the absence of this system, pathogenic Yersinia only cause local symptoms of moderate intensity.58
In general, Y. enterocolitica is primarily a gastrointestinal tract pathogen which, under defined host conditions, has a strong propensity for extra-intestinal spread. The clinical manifestations of the infection depend, to some extent, on the age and physical condition of the patient, the presence of any underlying medical conditions and the bioserotype of the organism.6 The most frequent occurrence, particularly in infants and children, is acute enteritis accompanied by fever, vomiting and inflammatory, occasionally bloody, watery diarrhea. The illness usually lasts from 3 to 28 days. However, in young adults the symptoms include acute terminal ileitis and mesenteric lymphadenitis with fever, diarrhea and abdominal pain usually localized in the right lower quadrant, mimicking appendicitis. This clinical syndrome usually lasts for 1–2 weeks. In more protracted cases of yersiniae, fatal necrotizing enterocolitis may occur, as well as a “pseudo-tumorigenic” form of suppurative mesenteric adenitis.1,20
Sepsis is a rare complication of Y. enterocolitica infection, except in immunocompromised patients, or those who have a predisposing underlying disease or are in an iron-overloaded state. However, it may also occur in normal hosts. The clinical course of septicemia may include abscess formation in the liver and spleen, pharyngitis, pneumonia, septic arthritis, meningitis, cellulitis, empyema and osteomyelitis, and may evolve into endocarditis or localize in the endovasculature of major blood vessels, leading to a mycotic aneurysm. In the setting of iron overload, intrinsically low-virulence Y. enterocolitica serogroups O:3 and O:9 may achieve virulence equal to that of serogroup O:8, since iron is easily provided in the environment without the necessity of the HPI-encoded iron capture system.1 Otherwise, acquisition of the infecting strain may also be associated with blood transfusion. Unfortunately, Y. enterocolitica has emerged as a significant cause of transfusion-associated bacteremia, and mortality occurs due to its ability to survive and replicate at low temperatures, even at 4°C. Despite this infection route being uncommon, when such an event occurs, the morbidity and mortality per individual may be significant. These results suggest that a transient, even occult bacteremia, may occur in a sub-population of individuals with Y. enterocolitica gastrointestinal infection. Long-term sequelae resulting from bacteremia can occur within a few weeks of the acute phase, with reactive arthritis and erythema nodosum being the most common. These post-infection manifestations are mainly seen in young adults and they have sometimes been reported to be particularly associated with serogroup O:3.1,6
Antimicrobial treatment and resistanceThe great majority of the gastrointestinal infections are self-limiting and confined to the gut and do not merit antimicrobial therapy in an immunocompetent host. However, antimicrobial therapy is warranted to treat enterocolitis in compromised hosts and in patients with septicemia or invasive infection, in which the mortality can be as high as 50%. Despite antibiotic susceptibility patterns varying among serogroups, the organism is usually susceptible in vitro to aminoglycosides, cotrimoxazole, chloramphenicol, tetracycline, third generation cephalosporins and fluoroquinolones but is resistant to penicillin, ampicillin and first generation cephalosporins. The intrinsic resistance to these beta-lactam antibiotics is due to the production of two chromosomally encoded beta-lactamase genes, blaA and blaB, encoding for one class A enzyme showing constitutive expression and one inducible class C enzyme (AmpC-type), respectively.83,84 The differential expression and activities of these two enzymes determines the level and spectrum of resistance. For example, both are predominantly produced among isolates belonging to serogroups O:3 and O:9.1,85 More recently, in biotype 1B strains, BlaA is involved in limiting the susceptibility to penicillins and cephalosporins whereas the BlaB contribution is primarily related to cephalosporin susceptibility.83
The WHO recommendations for antimicrobial chemotherapy initially included tetracycline, chloramphenicol, gentamicin and cotrimoxazole. More recently, newer compounds such as third generation cephalosporins and fluoroquinolones, which have excellent in vitro activity, have been considered as better alternatives.85,86 Their introduction has led to a significant decrease in the percentage of mortality due to septicemia. The use of fluoroquinolones has been particularly associated with a higher cure rate and a better response to fever. Nonetheless, several case reports of failure or poor response with cephalosporin treatment have been described despite their in vitro efficacy.85
Thus far, fluoroquinolones have been successfully used to treat liver abscess and pericarditis in addition to septicemia. Moreover, there is evidence that patients with reactive arthritis treated with ciprofloxacin (second-generation quinolone) show an earlier relief of pain and faster remission,85,87 despite there being other studies that suggest no benefit is observed or that increased fecal excretion may occur upon late-onset therapy in rats.88,89 A possible explanation for this greater success in comparison with cephalosporins may account for its excellent in vitro activity, superior tissue penetration and intracellular activity. Thus, ciprofloxacin should be considered as the first line agent for treating invasive infections due to Y. enterocolitica.85 Unfortunately, the levels of resistance to nalidixic acid (first-generation quinolone) are increasing: a percentage as high as 23% was detected in strains isolated in 2002 in Spain90 in comparison with the reduced 5% of resistance during the period 1995–2000.91,92 According to the mechanisms of resistance to quinolones described for Y. enterocolitica (90,93, and see review for general mechanisms of quinolone resistance94), this phenotype may lead to clinical failures when administering ciprofloxacin to treat Yersinia infections. On the contrary, thus far there are studies showing no resistance to ceftriaxone or other third generation cephalosporins among Y. enterocolitica strains of human origin.95–97
Concluding remarksInfections caused by the pathogen Y. enterocolitica following the ingestion of contaminated foods or water, generally do not transcend the gastrointestinal tract and the underlying lymphoid tissues (PP and MLN). Nonetheless, there are particular situations in which these bacteria can circumvent the host defense mechanisms and cause systemic infection, reaching extra-intestinal sites such as the liver and the spleen. Otherwise, an alternative less frequent, although serious route, of infection is through blood transfusion. Several cases have been reported due to the ability of this pathogen to grow at 4°C. Thus, although Y. enterocolitica is not among the most frequent agents causing human infection, the disease triggered can be life-threatening and relies on the presence of several virulence factors which allow the bacteria to subvert the host barriers and persist for a long time within the human host. Some of these virulence packages, however, cannot be detected along all the biotypes, but, when present, they have been shown to correlate with increased virulence such as that reported for biotype 1B strains (positive for two additional pathogenicity islands, HPI and Ysa).
The clinical features usually refer to acute diarrhea, fever, abdominal pain and pseudo-appendicitis. Nonetheless, more serious complications can also occur upon systemic dissemination, such as reactive arthritis and erythema nodosum. Currently, fluoroquinolones, as well as third generation cephalosporins, are the most appropriate treatments, when necessary, for combating such infections. Unfortunately, despite the susceptibility to these broad-spectrum beta-lactams remaining favorable, increased levels of resistance to nalidixic acid (a first-generation quinolone) among Y. enterocolitica clinical isolates are emerging. Therefore, the efficacy of the antimicrobial therapy may be compromised as a consequence of the increasing phenotype of decreased susceptibility to fluoroquinolones. Moreover, subsequent dissemination from other enteric bacteria of plasmid-encoded determinants conferring resistance to third-generation cephalosporins cannot be ruled out.
FundingThis study has been supported by the Spanish Ministry of Health (FIS 08/0195 to JV), by 2009 SGR 1256 from the Departament de Universitats, Recerca i Societat de la Informació de la Generalitat de Catalunya, and by the Ministerio de Sanidad y Consumo, Instituto de Salud Carlos III, Spanish Network for the Research in Infectious Disease (REIPI 06/0008). This work has also been supported by funding from the European Community (AntiPathoGN contract HEALTH-F3-2008-223101; TROCAR contract HEALTH-F3-2008-223031).
Conflict of interestThe authors declare no conflicts of interest related to this study.

