




The rapid identification and detection of carbapenemase-producing Klebsiella pneumoniae (CPKP) isolates is crucial to ascertain outbreaks, as well as to limit their spread. The current reference method for this purpose is multilocus sequence typing (MLST), which is laborious and expensive. Consequently, alternative typing methods are gaining attention, such as Matrix-Assisted Laser Desorption Ionization–Time Of Flight Mass Spectrometry (MALDI-TOF MS).
MethodsThis study sought to analyze MALDI-TOF MS as a typing method using 44 CPKP isolates that were well characterized by MLST. The most common types of samples from which these pathogens were isolated were skin and soft tissues (32%) and urine (29%). Half of the CPKP isolates were from hospitalized patients. Two approaches were followed for the analysis of the mass peak data obtained by MALDI-TOF MS. The first using all peaks obtained and the second using a selection of 21 characteristic peaks.
ResultsThe selection of 21 characteristic peaks showed greater discrimination power for ST11 and ST101. Principal component analysis (PCA) indicated that this dataset could be efficiently grouped with lineal classifiers. A Support Vector Machine (SVM) was chosen for this purpose after checking its capacity to classify bacterial strains on the basis of MALDI-TOF MS information.
ConclusionSVM was able to discriminate between ST11 and ST101 with high accuracy. In conclusion, our results reveal MALDI-TOF MS as a promising alternative technique for typing of CPKP isolates.
La rápida identificación y detección de los aislados de Klebsiella pneumoniae productores de carbapenemasas (CPKP) es crucial para identificar brotes e impedir la propagación de los aislados resistentes. El método de referencia para este propósito es el multilocus sequencing typing (MLST), que es un técnica laboriosa y cara, por lo que se buscan métodos de tipado alternativos que pueden desempeñar la misma función con menor esfuerzo. Entre las posibles técnicas se encuentra la espectrometría de masas de tiempo de vuelo MALDI-TOF.
MétodosEste estudio se han utilizado el sistema MALDI-TOF MS para tipar 44 aislamientos de CPKP previamente caracterizados por MLST. Las muestras clínicas de las que proceden los aislados son principalmente piel y tejidos blandos (32%) y orina (29%). La mitad de los aislamientos de CPKP procedían de pacientes ingresados. El análisis los datos obtenidos por MALDI-TOF MS se realizó con 2 enfoques diferentes, el primero usando todos los picos obtenidos y el segundo usando una selección de picos.
ResultadosLa selección de 21 picos característicos ofreció un mayor poder de discriminación entre ST11 y ST101. El análisis de componentes principales (PCA) indicó que este conjunto de datos podría agruparse eficientemente con clasificadores lineales. Para realizar este agrupamiento se escogió el algoritmo support vector machine (SVM, máquinas de vectores de soporte) para este propósito después de verificar su capacidad para clasificar las cepas bacterianas en base a la información de MALDI-TOF MS.
ConclusiónSVM pudo discriminar entre ST11 y ST101 con alta precisión. En conclusión, nuestros resultados revelan MALDI-TOF MS puede ser una técnica alternativa para el tipificación de aislamientos de CPKP.
The clonal expansion of multi-resistant isolates is a major public health problem. An example is reflected by carbapenemase-producing Klebsiella pneumoniae (CPKP), which are a cause of different types of infections for which there are limited treatment options. The rapid detection and typing analysis of such isolates is required in order to detect outbreaks.1,2 Even though multiple typing methods are available, the standard reference technique is Multilocus Sequence Typing (MLST).3 This is a DNA sequence-based typing method where 7 housekeeping genes are analyzed.4 Although MLST allows comparisons worldwide, it is not feasible for routine analysis in clinical microbiological laboratories due to its cost and complexity. Therefore, other typing techniques have emerged, such as Matrix-Assisted Laser Desorption Ionization–Time Of Flight Mass Spectrometry (MALDI-TOF MS). MALDI-TOF MS has been successfully applied for the rapid identification of bacteria and yeasts.5 Indeed, the number of studies supporting the value of MALDI-TOF MS in bacterial epidemiology is increasing.6,7 However, the results obtained with MALDI-TOF MS do not always correlate with those of MLST.8,9 It should be taken into account that the former classifies bacteria based on functional and expressed proteins while the MLST use core genome (house-keeping genes) for doing the dendrogram. Thus, MALDI-TOF MS is especially useful for identifying bacteria associated with a specific phenotype and therefore with a specific area. In this sense, MS biotyping can be used to determine the geographical origin of unknown pathogenic isolates.
MALDI-TOF-MS has the advantage over MLST in that it is easier to perform and quicker. However, the most limiting factor for the typing of microorganisms by MALDI-TOF is reproducibility, so a good standardization for working conditions is required.
The aim of the present study was to evaluate different algorithms for the correct classification of protein spectra from different K. pneumoniae clones and validate the most suitable ones prior to their implementation for rapid bacterial typing with MALDI-TOF.
Materials and methodsBacterial strains and reference typing (MLST)Forty-four non-replicate CPKP isolates were recovered from clinical samples. Isolates were collected from September 2010 to October 2013. The isolates were collected from the following: urine (n=13; 29%), lower respiratory tract (n=10; 23%), skin and soft tissues (n=14; 32%) and various other sites (n=7; 16%). Twenty-two isolates were from patients in medical wards (50%), 12 from patients in the intensive care unit (27%), 6 from patients in surgical wards (14%), and 4 from outpatients (9%). Isolates were identified by MALDI-TOF-MS. Carbapenemase production was screened by the modified CIM test, and carbapenemase activity was inhibited using the combined disk test EDTA and phenylboronic acid, as previously described.10 Briefly, inhibition zones obtained by using meropenem disks with or wihtout EDTA (10μl of a 0.5M solution) and phenylboronic acid (400μg) were analyzed and compared. PCR was used for the detection of the carbapenemase genes blaKPC, blaVIM, and blaOXA-48-like.
Typing by MLST showed 10 sequence types (STs), namely ST11 (19 isolates), ST101 (13 isolates), ST15 (3 isolates), ST405 (2 isolates), ST307 (2 isolates), and ST431, ST578, ST198, ST323 and ST37 (all with 1 isolate each). The presence of carbapenemase genes in the analyzed clones was as follows: 1 IMP-22 belonged to ST578; 3 OXA-48-like to ST307 (2 isolates) and to ST11 (1 isolate); 13 KPC-2 to ST101; and 27 VIM-1 to the remaining STs.
Bacterial typing using MALDI-TOF MS (Bruker Daltonics, Bremen, Germany)A McFarland 3.0 bacterial suspension was prepared in 70% ethanol. After centrifugation, the pellet was resuspended in 70% formic acid and pure acetonitrile. Then, 1μL of the supernatant was spotted onto the MALDI target plate. Finally, the spots were overlayed with 1μL of matrix solution HCCA (saturated solution of α-cyano-4-hydroxycinnamic acid matrix) in 50% acetonitrile and 2.5% trifluoroacetic acid solution.
MALDI spectra for each strain were acquired in the same experiment using a Microflex LT controlled by FlexControl software (Bruker Daltonik, Germany) in linear positive ion mode with a mass range of 2–20kDa. For each sample, 8 applications were performed onto the MALDI target and 3 spectra of each sample spot were automatically acquired. A total of 240 of laser shots were used to acquire each spectrum for a total of 5760 per sample. Spectra were externally calibrated by using the BTS (Bacterial Test Standard) and subsequently analyzed with the software FlexAnalysis v3.3 (Bruker Daltonik, Germany).
Peaks were selected using a centroid algorithm with a S/N ratio of 2 and a minimum intensity threshold of 300. The preprocessing included smoothing, baseline subtraction (MBT_Standard.FAMSMethod), and internal peak alignment. Spectra were aligned by internal recalibration using BTS mixed with K. pneumoniae extract.
We tested two approaches to classify the 44 CPKP isolates: one using all the peaks obtained, and other using a selection of the same. For this purpose, MALDI Biotyper 3.0 (BrukerDaltonics, Germany) and SPSS software were used to produce dendrograms. With the Biotyper, MSP dendrograms were obtained with a consensus spectrum using the conditions established by the manufacturer (correlation distance measured with the average linkage algorithm) and using correlation distance measured with Ward's linkage. Clusters were analyzed on the basis of arbitrary distances of 700 and 200 for average linkage and at 500 and 100 for Ward's linkage. Before analysis by SPSS software, a list of peaks for each isolate was exported from FlexAnalysis to Microsoft Excel. For each sample, a consensus spectrum with the peaks obtained in the 24 spectra was obtained. The average peak and the number of times that it appeared in these spectra were considered. A selection of characteristic peaks was made by visual examination. Although visual inspection is somewhat subjective and can make it difficult to transfer to the routine, the selected peaks were clear enough to be the ones selected for all the authors.
Dendrograms (or phylogenetic tree) produced by SPSS were based on hierarchical cluster analysis using squared Euclidean distance measured by Ward's linkage. Arbitrary cut-offs of 5% and 20% were used to build this branching diagram.
In addition, a Principal Components Analysis (PCA) and a classification with the Support Vector Machine (SVM) were carried out. PCA is a technique able to replace the original variables by smaller number of derived variables, namely the principal components (PCs), which are linear combinations of the original ones.11 PCA was performed for the most common clones: ST11 and ST101. The dataset was projected in two dimensions defined by the first and second PC and the second and third PC using all the peaks and 21 characteristic peaks (selected by visual examination) respectively.
SVM, a binary classifier and present in the Python framework Scikit-learn, was used to classify the CPKP isolates into three groups, ST11, ST101 and the remaining STs, using peak data as input. SVM is a discriminative classification algorithm that has been shown to be very successful for biological applications.12 Isolates were binned on the basis of the accuracy of the experiment, and each bin was labeled with the percent of times it appeared, while outputs were a labeled vector corresponding to ST11, ST101, or the rest of the STs. In order to prevent overfitting, the SVM was trained in a subset of the data (training set) and subsequently evaluated against the other, unseen data (test set). The algorithm was run 50 times, each time taking a random 70% of the data as the training set and the rest as the test set. In order to measure the replicability of the experiment, we repeated it three times.
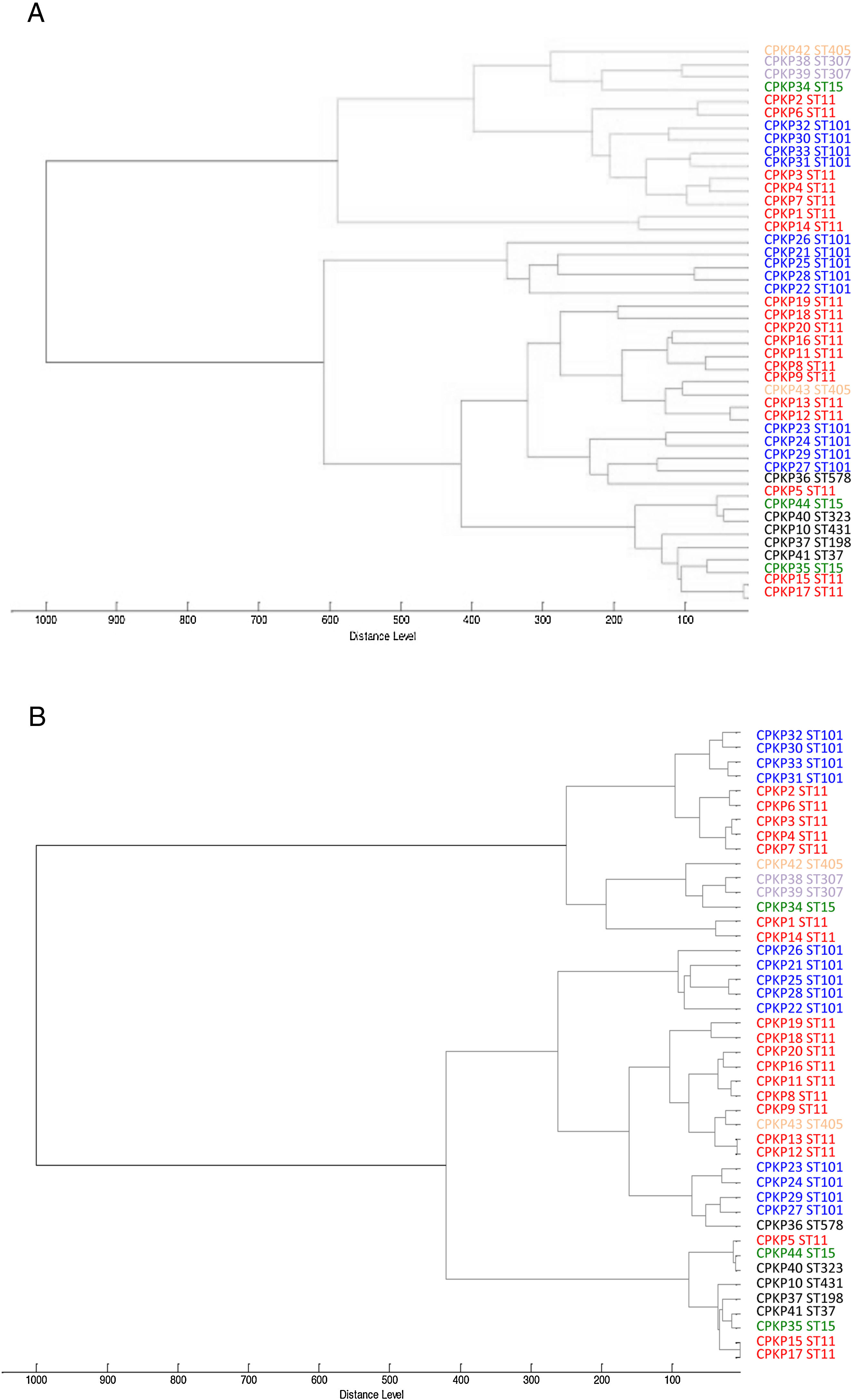
ResultsAfter obtaining the profile of the peaks of the 44 CPKP isolates and processing them by Biotyper, we obtained the dendrograms shown in Fig. 1a (average linkage) and 1b (Ward linkage) using all peaks. In both dendrograms, using a cut-off for distance of 700 by average linkage and 500 by Ward linkage, all isolates were grouped into 2 clusters, these comprising a mixture of STs. Mostly, these groups coincided in the 2 dendrograms, although some strains were linked in a different way. With a lower cut-off (200 by average linkage and 100 by Ward linkage), there were several clusters with more homogeneity in the grouping of STs. Using these cut-offs, the 2 CPKP isolates belonging to ST307 were grouped in the same cluster. However, the two isolates belonging to ST405 were grouped in a different cluster. Only 2 out of 3 isolates belonging to ST15 were grouped together. Focusing on the two major clones, both dendrograms showed two major clusters in which ST11 and ST101 were linked in similar form. Isolates from both STs were included in several clades or sub-clades in which isolates from other minority STs were also included.
 Average linkage was used. (B) Ward linkage was used.")
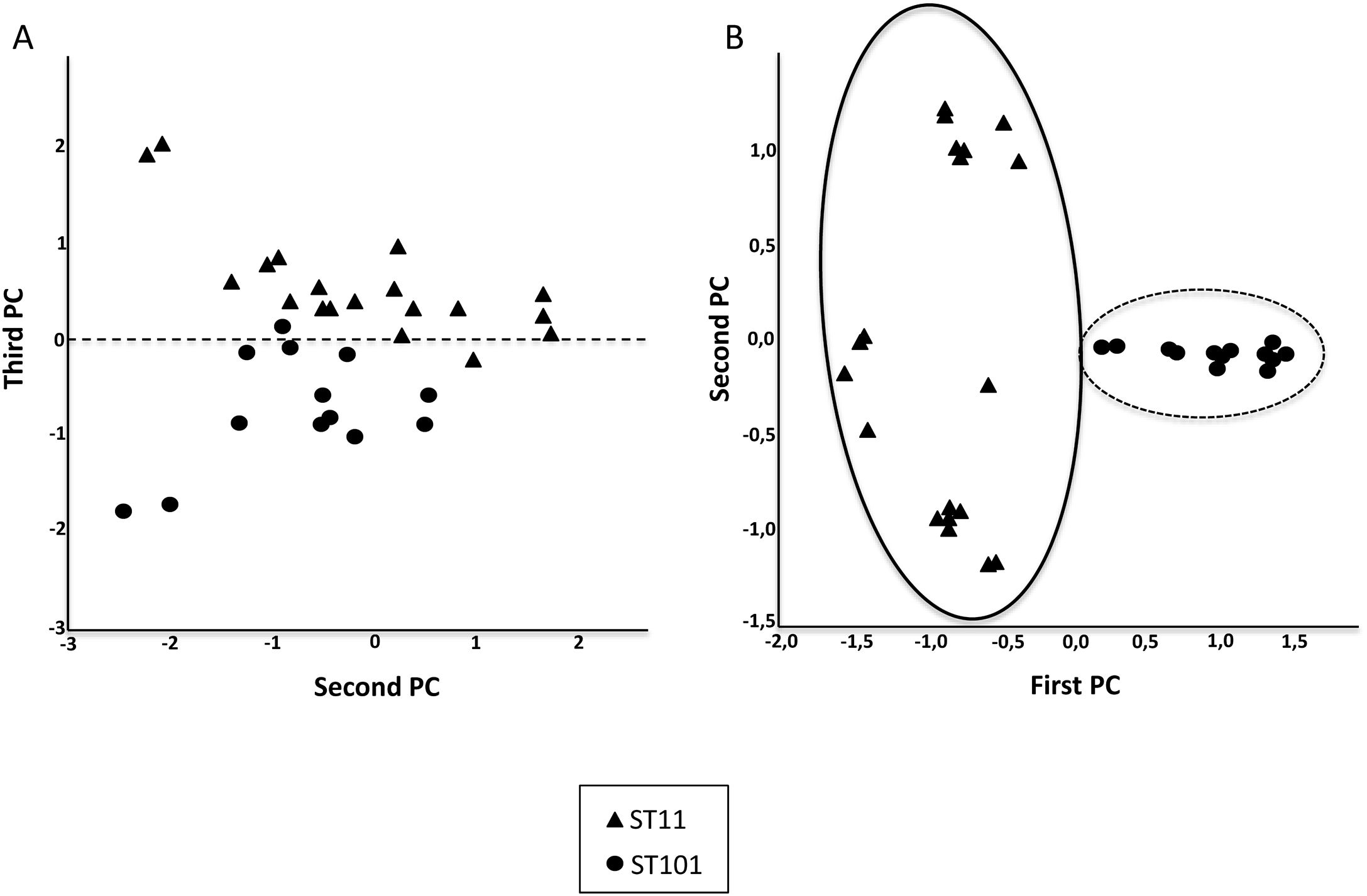
Fig. 2 shows the 32 CPKP isolates of the main STs in the axis provided by the PCA. PC2 and PC3 showed a high capacity to discriminate between ST11 and ST101. Drawing a line at PC1=0, isolates with ST11 were grouped on one side (negative values) and those with ST101 on the other (positive values).
 and ST101 (circle). (A) All peaks were used. (B) Twenty-one peaks were used.")
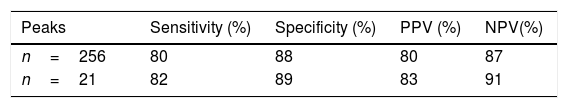
After analysis and comparison of the dendrograms, the SVM algorithm was applied to determine whether it improved the agreement with the results obtained by MLST. Its values of sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) were 80%, 88%, 80% and 87%, respectively (Table 1). Two of the 13 isolates belonging to ST101 and 4 of the 19 belonging to ST11 were not correctly classified.
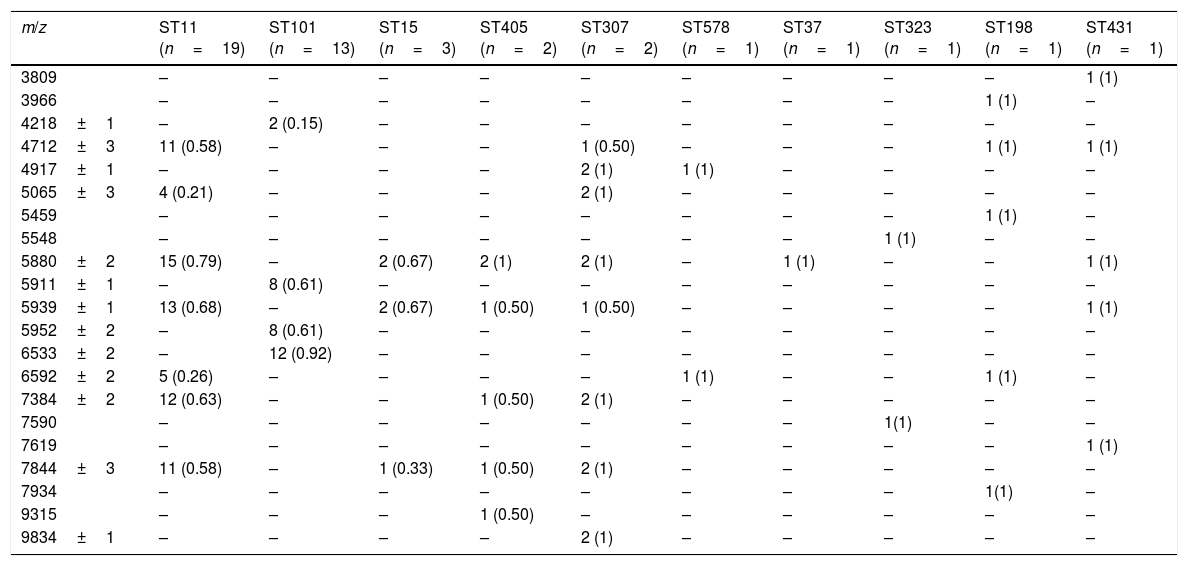
Twenty-one characteristic peaks were selected to discriminate between STs (Table 2). Although each of the main STs had a unique set of peaks, this was not the case when minority STs were added, as reflected by the observation that ST11 showed shared peaks.
Identifying peaks of the different clones.
| m/z | ST11 (n=19) | ST101 (n=13) | ST15 (n=3) | ST405 (n=2) | ST307 (n=2) | ST578 (n=1) | ST37 (n=1) | ST323 (n=1) | ST198 (n=1) | ST431 (n=1) |
|---|---|---|---|---|---|---|---|---|---|---|
| 3809 | – | – | – | – | – | – | – | – | – | 1 (1) |
| 3966 | – | – | – | – | – | – | – | – | 1 (1) | – |
| 4218±1 | – | 2 (0.15) | – | – | – | – | – | – | – | – |
| 4712±3 | 11 (0.58) | – | – | – | 1 (0.50) | – | – | – | 1 (1) | 1 (1) |
| 4917±1 | – | – | – | – | 2 (1) | 1 (1) | – | – | – | – |
| 5065±3 | 4 (0.21) | – | – | – | 2 (1) | – | – | – | – | – |
| 5459 | – | – | – | – | – | – | – | – | 1 (1) | – |
| 5548 | – | – | – | – | – | – | – | 1 (1) | – | – |
| 5880±2 | 15 (0.79) | – | 2 (0.67) | 2 (1) | 2 (1) | – | 1 (1) | – | – | 1 (1) |
| 5911±1 | – | 8 (0.61) | – | – | – | – | – | – | – | – |
| 5939±1 | 13 (0.68) | – | 2 (0.67) | 1 (0.50) | 1 (0.50) | – | – | – | – | 1 (1) |
| 5952±2 | – | 8 (0.61) | – | – | – | – | – | – | – | – |
| 6533±2 | – | 12 (0.92) | – | – | – | – | – | – | – | – |
| 6592±2 | 5 (0.26) | – | – | – | – | 1 (1) | – | – | 1 (1) | – |
| 7384±2 | 12 (0.63) | – | – | 1 (0.50) | 2 (1) | – | – | – | – | – |
| 7590 | – | – | – | – | – | – | – | 1(1) | – | – |
| 7619 | – | – | – | – | – | – | – | – | – | 1 (1) |
| 7844±3 | 11 (0.58) | – | 1 (0.33) | 1 (0.50) | 2 (1) | – | – | – | – | – |
| 7934 | – | – | – | – | – | – | – | – | 1(1) | – |
| 9315 | – | – | – | 1 (0.50) | – | – | – | – | – | – |
| 9834±1 | – | – | – | – | 2 (1) | – | – | – | – | – |
Nevertheless, four specific biomarkers remain exclusive to ST101 with peaks at 4218, 5911, 5952 and 6533 m/z. The last of the mentioned peaks was observed in all except one of ST101 isolates, but in none of the other STs.
ST11 showed common peaks with minority STs, but the association of present/absent peaks was still unique to each of the STs analyzed. The greatest coincidences were with ST307, but the presence of peaks 4917 and 9834 m/z in the latter, could be used to correctly distinguish between both groups of isolates.
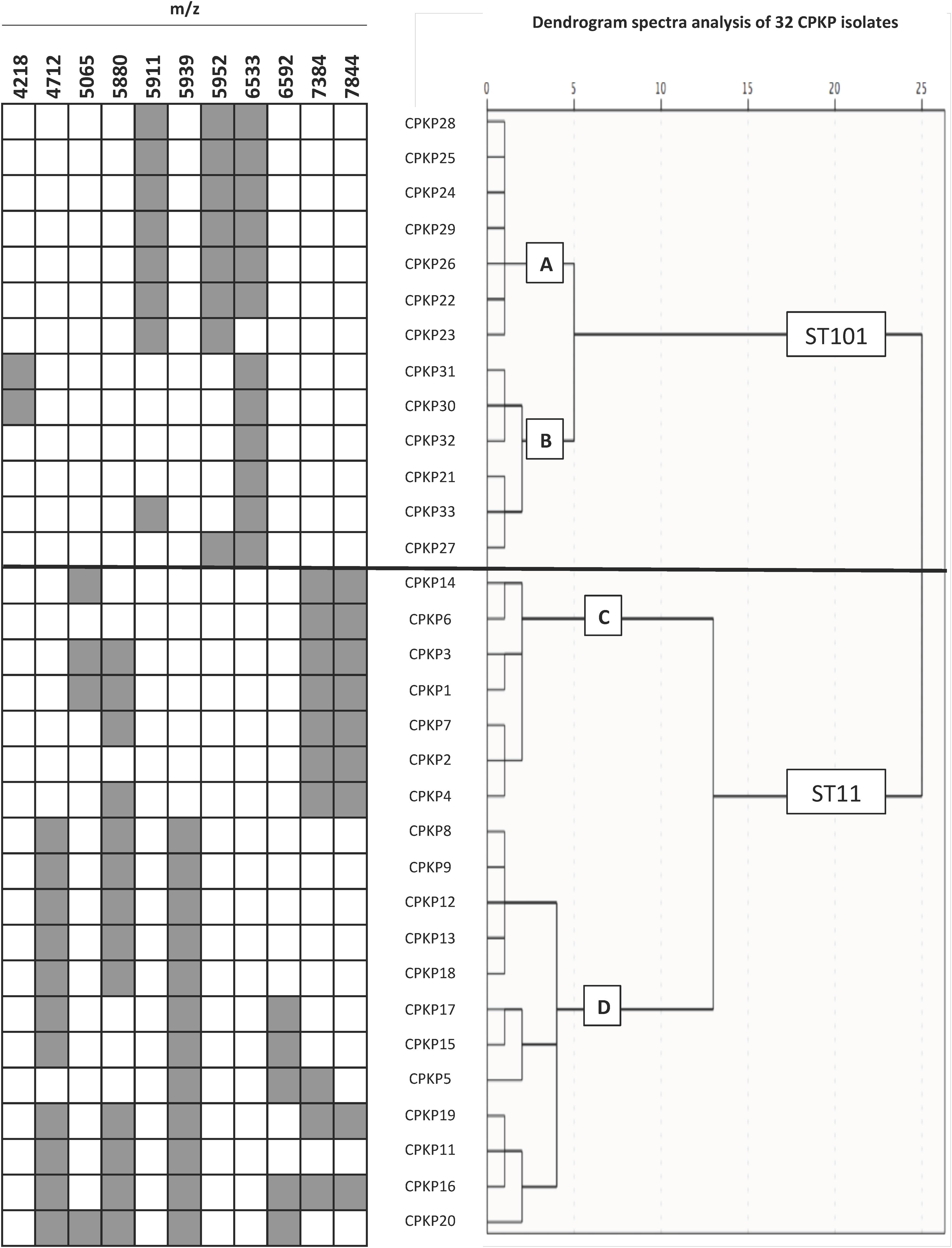
To confirm the ability of the selected peaks to distinguish clones, a dendrogram using SPSS software was produced using squared Euclidean distance and Ward linkage. Using an arbitrary cut-off of 20%, ST11 and ST101 were completely separated, and as expected, and minority STs were mixed in with the ST11 cluster. Fig. 3 shows a dendrogram of 32 isolates (ST11 and ST101) with the characteristic peaks for each ST. Using a cut-off of 5%, 2 groups with the same pattern of peaks was detected for each ST.
 using 21 peaks were represented with the characteristics peaks of each ST.")
After observing that the clustering of the CPKP strains improved with peak selection, we sought to verify that PCA and the SVM algorithm also enhanced the clustering. In this regard, both approaches contributed to enhance clustering (Fig. 2b and Table 1).
DiscussionMLST is a method used to type bacterial DNA that analyzes 7 housekeeping genes. MALDI-TOF MS analyzes ribosomal and surface-expressed proteins (a small proportion of all proteins expressed by bacteria). Since each of these two methods focuses on a different property, they will not necessarily cluster strains the same way.
MALDI-TOF MS has been used for cluster analysis of several microorganisms, such as Streptococcus pyogenes,13Enterococcusfaecium14 and Listeria monocytogenes.15 Furthermore, this mass spectrometry technique has proven capacity to discriminate major S. aureus clonal complexes.16–18 In addition, this method has high resolution for ESBL-producing E. coli isolates, since it can distinguish the ST131 clonal group from others.19 Nevertheless, regarding the typing of K. pneumoniae and Acinetobacter baumannii isolates by MALDI-TOF MS, the majority of previous studies did not compare the performance of MLST and MALDI-TOF MS9,10,20 or did it, but found that MALDI-TOF MS is not able to properly discriminate different clusters.21,22 It should be remembered that the Biotyper system developed by Bruker is mainly focused on the identification of microorganisms at the species level. Typing does not lie within the scope of this system and therefore, their clustering method has many flaws.
It should be noted that peak data obtained by MALDI-TOF MS could be processed using various software packages and algorithms. In this regard, several authors have applied different machine learning (ML) algorithms, such as Support Vector Machine, to identify bacterial clones.23,24 SVM is a method of conducting learning algorithms that analyze data used for classification and regression analysis.
Consequently, there is great diversity in the classification of bacterial strains, thereby hindering comparison of results between studies. Here we analyzed two approaches on the basis of the number of peaks used for the classification of 44 CPKP isolates. First, using all peaks obtained in the spectrum, we sought to examine how Biotyper clustered the strains. For this purpose, we used two linkages: average and Ward's. Although the grouping of the isolates was quite similar, the best clustering was achieved by using Ward's linkage: clusters were clearer and the distance measures were lower. Using this linkage algorithm, the clustering was optimal because the groups formed did not distort the original data. Ward's algorithm is one of the most widely used linkage methods. It has virtually all the advantages of average linkage and usually shows greater discrimination power to determine clustering levels.
Analysis of mass data of the strains belonging to ST11 and ST101 with PCA revealed a separation between the two clones. Based on this observation, the SVM algorithm was applied to classify the CPKP strains, presenting a high sensitivity and specificity. Dendrograms separated ST11 and ST101; however, SVM was also able to group strains of the same clone. These results contrast with those reported by Angeletti et al., whose algorithms separated CRPK strains with the same ST into two clusters.25
After visual examination of mass peak data of each isolate, we identified 21 characteristic peaks of the different STs, of which 11 peaks separated isolates of the main STs (ST11 and ST101). We propose that these peaks could serve as biomarkers to identify K. pneumoniae isolates belonging to these clones. In addition, better results for PCA analysis and the SVM algorithm were achieved when using the 21 characteristic peaks than when using all peaks. Compared to previous studies,16,17 the sensitivity and specificity of our algorithm were not as high. These differences could be attributed to the number of isolates, which in our case was considerably lower.
Statistical programs, such as ClinProTools, which are specifically designed to process peak data obtained by MALDI-TOF MS, generate pattern recognition models. Although ClinProTools provides satisfactory results,16,17 its software is not included in the standard package and it is not freely available to some laboratories. Since MALDI Biotyper software is not able to produce dendrograms on the basis of only certain peaks, general statistical programs like SPSS software can be used. Such programs have the drawback of being more laborious with respect to data processing than ClinProTools. For each sample, we had to manually create a consensus spectrum with the peaks obtained in the 24 spectra, since it is not possible to export this information from Biotyper. However, MALDI Biotyper software provides an alternative method for processing peak data when the ClinProTools software is not available.
In conclusion, our data suggest that MALDI-TOF MS can be a useful tool for typing CPKP isolates. Although our results are slightly biased because the number of samples belonging to ST11 and ST101 is much higher than these belonging to other STs, they indicate that MALDI-TOF MS has the ability to classify CPKP strains on the basis of MLST criteria. Of the two approaches analyzed, the selection of characteristic peaks had the greatest power of discrimination between ST11 and ST101. Eleven peaks identified ST11 and ST101, thereby supporting their utility as potential biomarkers of these clones. Finally, we propose an alternative method for analyzing peak data by means of more accessible programs. Since the results with MALDI-TOF MS showed good correlation with those obtained by MLST, a database with the main CPKP clones circulating in each hospital could be built, thus facilitating the rapid identification of an outbreak. However, further studies with a greater number of CPKP isolates are required.
Transparency declarationsNone to declare.
Conflict of interestThe authors declare that they have no conflict of interest.
