






Generar una secuencia consenso a partir de los datos de secuenciación masiva obtenidos en estudios de resistencias a antiretrovirales, que sea representativa de la secuencia Sanger y que sirva para estudios de epidemiología molecular.
Material y métodosEn 62 pacientes se obtuvo la secuencia de transcriptasa reversa-proteasa, mediante Sanger (Trugene-Siemens), y NGS (454GSJunior-Roche). Las secuencias consenso NGS se generaron con Mesquite, seleccionando umbrales 10%, 15% y 20%. Para el estudio filogenético se empleó MEGA.
ResultadosUtilizando el umbral 10%, 17/62 pacientes presentaron secuencias pareadas NGS-Sanger, con una mediana de bootstrap del 88% (IQR83,5-95,5). La asociación aumenta a 36/62 pacientes y el bootstrap, a 94% (IQR85,5-98), y alcanza el máximo al 20% en 61/62 pacientes, bootstrap 99% (IQR98-100).
ConclusiónMostramos un método seguro para generar secuencias consenso NGS para su uso en estudios de epidemiología molecular procesadas con umbral 20%, de fácil uso y aplicación en los servicios de microbiología clínica.
To show how to generate a consensus sequence from the information of massive parallel sequences data obtained from routine HIV anti-retroviral resistance studies, and that may be suitable for molecular epidemiology studies.
Material and methodsPaired Sanger (Trugene-Siemens) and next-generation sequencing (NGS) (454 GSJunior-Roche) HIV RT and protease sequences from 62 patients were studied. NGS consensus sequences were generated using Mesquite, using 10%, 15%, and 20% thresholds. Molecular evolutionary genetics analysis (MEGA) was used for phylogenetic studies.
ResultsAt a 10% threshold, NGS-Sanger sequences from 17/62 patients were phylogenetically related, with a median bootstrap-value of 88% (IQR83.5-95.5). Association increased to 36/62 sequences, median bootstrap 94% (IQR85.5-98)], using a 15% threshold. Maximum association was at the 20% threshold, with 61/62 sequences associated, and a median bootstrap value of 99% (IQR98-100).
ConclusionA safe method is presented to generate consensus sequences from HIV-NGS data at 20% threshold, which will prove useful for molecular epidemiological studies.
Recientemente, un buen número de servicios de microbiología clínica han adoptado las técnicas de secuenciación masiva (next generation sequencing [NGS]) para los estudios de resistencias a antirretrovirales en pacientes VIH. La capacidad de NGS en la detección de variantes virales de baja frecuencia se ha determinado en varios estudios1, disminuyendo la sensibilidad en la detección de mutaciones de resistencia hasta niveles del 1% (variantes minoritarias), lo que proporciona ventajas para la elección de la mejor línea de tratamiento y evitar el fracaso al tratamiento2,3. En nuestro país, uno de los motivos de la instauración de NGS para la detección de resistencias a antirretrovirales ha sido la discontinuación de los métodos de secuenciación Sanger comerciales por alguno de los proveedores.
Las secuencias de proteasa (PR) y transcriptasa reversa (RT) obtenidas de los ensayos para determinar resistencias se utilizan a menudo por parte de investigadores en estudios de epidemiología molecular, mediante el empleo de técnicas de filogenética y filodinámica4. Con la introducción de las técnicas de NGS, esta información se puede perder debido a que el manejo y el almacenamiento de las secuencias para este tipo de estudios son complejos; además, si las secuencias de NGS no se tratan apropiadamente, pueden aportar resultados equivocados. Para emplear las secuencias de NGS en estudios filogenéticos se requiere tanto una formación especial para el procesado de secuencias, como de ordenadores de gran potencia para procesar el gran volumen de datos obtenidos5. Para los estudios de epidemiología molecular, una alternativa es generar una única secuencia consenso de NGS, pero algunos estudios no son claros u omiten el método utilizado para generarla6; además, no conocemos con certeza cuál es la representatividad de esta consenso de NGS de la secuencia obtenida por Sanger, y cómo influyen los puntos de corte que utilicemos para generar dicho consenso.
El objetivo de nuestro trabajo ha sido determinar cuál es el mejor umbral de corte para la obtención de una secuencia consenso NGS que sea representativa de la secuencia tipo Sanger y que pueda ser utilizada en estudios de epidemiología molecular.
MétodosPara nuestro estudio hemos utilizado secuencias de 62 pacientes naïve del periodo 2014-2015, nuevos diagnósticos VIH, referidos para estudios de resistencias a antirretrovirales. Las secuencias tipo Sanger se obtuvieron utilizando Trugene®HIV-1Genotyping (Siemens-[NAD]). Para NGS utilizamos el kit GSVType HIV-1 Drug Resistance Primer (Roche) para 454 GS-Junior, partiendo del mismo ARN. Las secuencias consenso de NGS se generan mediante el software Mesquite v. 2.75, seleccionando umbrales de corte del 10, del 15 y del 20%. Previo a la utilización de Mesquite se efectúa un filtrado de las secuencias, utilizando los comandos fastq_filter del software Usearch según longitud deseada de amplicón y calidad de secuencia (>30Q). Mesquite7 es un programa que funciona mediante iconos y pestañas, siendo intuitivo. Para su utilización es necesario exportar las secuencias filtradas en formato pfam y seleccionar el umbral de corte para la creación de la secuencia consenso, exportándola en formato fasta. Posteriormente, las secuencias del gen pol (PR 4-99; RT 38-247) se procesan, alinean mediante MUSCLE en MEGA 6.06 y se generan árboles filogenéticos mediante el método de máxima verosimilitud, utilizando el modelo General Time Reversible (GTR) para el cálculo de las distancias evolutivas, con una distribución gamma equivalente a 1,89, obtenido con FindModel DNA y utilizando remuestreo de bootstrap con 1.000 réplicas para construir los árboles filogenéticos consenso. Para definir una relación entre secuencias se tienen en cuenta solo las ramas pertenecientes a clusters con un valor de bootstrap superior al 75%. Finalmente, los árboles son procesados en FigTree v. 1.4.2. El análisis del subtipo viral se realizó utilizando REGA HIV-1Subtyping Tool v. 3.0.
ResultadosNuestro estudio ha incluido 62 pacientes VIH-1, naïve, mediana de edad de 37años (IQR30-45), carga viral (mediana) 74.900cp/ml (IQR20.715-176.250), recuento de CD4 (mediana) 430células/ml (IQR48,5-567,78); el 82% eran hombres.
Para evaluar la concordancia entre las secuencias consenso de NGS con diferentes umbrales y la secuencia original de Sanger hemos analizado el número de secuencias que se asocian por pares entre sí, y los valores de bootstrap entre los pares. Utilizando un umbral de corte al 10% se observa que solo en 17/62 (27%) pacientes las secuencias Sanger están pareadas con NGS de la misma muestra, y en estas, la mediana de bootstrap fue del 88% (IQR83,5-95,5). Aumentando el umbral al 15%, las secuencias se asocian por pares en 36/62 (58%) pacientes, con una mediana de bootstrap del 94% (IQR85,5-98). Al 20%, esto sucede en 61/62 pacientes con una mediana de bootstrap del 99% (IQR98-100) (fig. 1); para el caso en que la secuencia de NGS no se asocia con la secuencia de Sanger, detectamos un gran número de diferencias entre bases.
NGS-10%; B)NGS-15%, y C)NGS-20%. Los valores de bootstrap están asociados según el color de la gráfica, siendo una buena relación a partir de 70%.")
Representación de los árboles filogenéticos en FigTree v. 1.4.2, formados por las secuencias Sanger y secuencias NGS a los distintos umbrales: A)NGS-10%; B)NGS-15%, y C)NGS-20%. Los valores de bootstrap están asociados según el color de la gráfica, siendo una buena relación a partir de 70%.
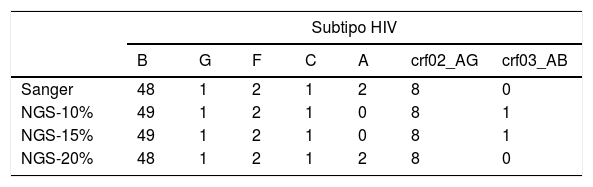
La mayoría de los pacientes estaban infectados por subtipoB (77,4%), seguido de CRF02_AG (12,9%), A y F (3,2%) y C y G (1,6%). Utilizando consenso NGS umbral 10% y 15% se observan 2 casos discordantes respecto al subtipo Sanger: un caso subtipo B-NGS y A1-Sanger, y otro desde subtipo CRF03_AB-NGS y A1-Sanger. Estas diferencias desaparecen al utilizar las secuencias consenso NGS umbral 20% (tabla 1). La figura 2 muestra la gráfica bootscan del subtipado en la segunda muestra discordante.
Distribución de subtipos virales HIV según las secuencias analizadas Sanger y secuencia consenso NGS a los distintos umbrales, mediante REGA HIV-1Subtyping Tool v. 3.0
| Subtipo HIV | |||||||
|---|---|---|---|---|---|---|---|
| B | G | F | C | A | crf02_AG | crf03_AB | |
| Sanger | 48 | 1 | 2 | 1 | 2 | 8 | 0 |
| NGS-10% | 49 | 1 | 2 | 1 | 0 | 8 | 1 |
| NGS-15% | 49 | 1 | 2 | 1 | 0 | 8 | 1 |
| NGS-20% | 48 | 1 | 2 | 1 | 2 | 8 | 0 |
, secuencia consenso NGS-10% (B) y NGS-20% (C), mediante REGA HIV-1Subtyping Tool v. 3.0. El bootscan ofrece un mismo valor de subtipado HIVA para la secuencia Sanger y secuencia consenso NGS-20%, sin embargo se observa un subtipado CRF03_AB para secuencia consenso NGS-10%.")
Bootscan de secuencia Sanger (A), secuencia consenso NGS-10% (B) y NGS-20% (C), mediante REGA HIV-1Subtyping Tool v. 3.0. El bootscan ofrece un mismo valor de subtipado HIVA para la secuencia Sanger y secuencia consenso NGS-20%, sin embargo se observa un subtipado CRF03_AB para secuencia consenso NGS-10%.
Los estudios filogenéticos en VIH8,9, en concreto los estudios de parentesco, dinámica de la epidemia VIH y de subtipado molecular utilizando la secuencia del gen pol, se han utilizado entre otros fines para conocer redes y nodos de transmisión del VIH, así como redes migratorias de los diferentes subtipos. Para estos objetivos la mayoría de los estudios publicados, a nivel internacional10 y a nivel local11,12, han utilizado la secuenciación de tipo Sanger. Algunos de estos estudios han utilizado toda la información obtenida mediante NGS13, pero por lo general se intenta generar una única secuencia consenso, habitualmente mediante comandos informáticos complejos. La transición desde la secuenciación Sanger a NGS para el estudio del gen pol en el análisis de mutaciones de resistencia ha provocado un cambio en el tipo de secuencias que manejamos en los servicios de microbiología clínica y paradójicamente puede suponer un freno para los estudios locales de epidemiología molecular de VIH en nuestro país. En nuestro trabajo proponemos la utilización de Mesquite, un software intuitivo, de fácil manejo y sin necesidad de comandos, que simplifica la obtención de la secuencia consenso a partir de secuencias obtenidas mediante NGS, demostrando que utilizando un umbral del 20% para generar esta consenso obtenemos una información segura, fiable y de idénticas características que la secuencia Sanger, que puede ser utilizada en estudios de epidemiología molecular, obviando la problemática actual de las secuencias obtenidas mediante NGS.
Como podemos observar en nuestro estudio, para poder utilizar con seguridad las secuencias consenso en estudios de epidemiología molecular en VIH, y para que la secuencia sea representativa de la secuencia tipo Sanger, debemos elevar el umbral de corte hasta el 20%. Solo así hemos conseguido una mediana de bootstrap del 99% (IQR98-100) entre las secuencias consenso de NGS y la tipo Sanger. Con umbrales del 10 o del 15% el porcentaje de secuencias que se asocian por pares NGS-Sanger y la mediana de bootstrap son insuficientes. Además, la variabilidad llega a ser tal que hasta en la asignación del subtipo viral se cometen errores, hecho que se corrige con el consenso al 20%. Estas discrepancias son debidas a la multitud de bases ambiguas generadas con umbral 10 y 15%, con una disminución del soporte estadístico para la correcta adjudicación del subtipo viral.
Una parte importante en los estudios de epidemiología molecular es el proceso de alineación de secuencias, teniendo como objetivo aproximar posiciones homólogas en base a la verdadera historia evolutiva de las secuencias14. El problema de la utilización de secuencias consenso NGS10% y 15% en tales estudios radica en la presencia de regiones ambiguas, presentando una incertidumbre sustancial, evitando la robustez de análisis estadísticos tanto filogenéticos15 como de subtipado, obteniendo resultados que no se corresponden a lo esperado.
Es importante indicar que la metodología que presentamos aquí es apropiada para obtener secuencias consenso para su uso en estudios de epidemiología molecular de VIH, pero no para el análisis de mutaciones de resistencia. La mayor sensibilidad de NGS para detectar variantes minoritarias y su utilidad clínica han sido estudiadas con detalle1-4. NGS proporciona una información muy valiosa respecto de la proporción relativa de una mutación con respecto al total de virus circulantes, información que se perdería al obtener la secuencia consenso.
En resumen, en nuestro trabajo presentamos una metodología que permite generar secuencias consenso que son representativas de la secuencia Sanger para su uso en estudios de epidemiología molecular, siendo necesario efectuar un procesado de las secuencias y utilizar puntos de corte de al menos el 20%.
FinanciaciónFondo de Investigación Sanitaria (PI12/01053, PI15/00713), RD12/0017/006 (Plan Nacional de I+D+I, Fondo Europeo de Desarrollo Regional-FEDER). Federico García disfruta de un Programa de Intensificación de la Actividad de Investigación del Servicio Andaluz de Salud. José Ángel Fernández-Caballero disfruta de un contrato de la RD12/0017/006.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

