


¿ INTRODUCCIÓN
A nivel global, se desconoce la incidencia real de los desórdenes linfoproliferativos postrasplante (PTLD, por sus siglas en inglés), debido a la imposibilidad para calcular la tasa ajustada de la enfermedad por el número de años de riesgo que presentan los pacientes afectos; no obstante, el número de casos nuevos se ha calculado en 224, 54 y 31 habitantes/año, durante el primero, segundo, y sexto años postrasplante, respectivamente.1 Esta condición parece ser relativamente común en los receptores de órganos sólidos, entre los cuales Burns y Crawford reportaron una frecuencia estimada de 10%.2 En los adultos trasplantados representa la segunda neoplasia en frecuencia después del cáncer de piel, mientras que en los niños, constituye la primera enfermedad tumoral en frecuencia. De forma similar, el PTLD significa la principal causa de muerte por cáncer en la población expuesta a algún trasplante de órgano sólido (la letalidad después del diagnóstico supera 50%).3,4
Varios estudios integrativos han caracterizado el comportamiento de la enfermedad en Estados Unidos y en Europa, encontrando que 85% de los PTLD tienen origen en las células B y que 80% de los casos están asociados con la infección por el virus de Epstein Barr (EBV).5 En contraposición, 15% de los PTLD provienen de las células con linaje T y NK, escenario donde sólo 30% tienen relación con la infección por este agente, siendo más relevante el contagio por el virus linfotrópico de las células T del humano (HTLV).6 A diferencia de los pacientes llevados a trasplante de médula ósea, en 95% de los sujetos con un injerto de órgano sólido la infección proviene de los linfocitos tumorales del receptor de órgano.7 Por otro lado, están los casos de PTLD en pacientes negativos para la infección por EBV; este subgrupo comprende entre 8% y 28% de los casos y se caracteriza por la presentación tardía (la mediana para el diagnóstico se encuentra entre los 17 y 174 meses postrasplante). En estos pacientes, la oncogénesis está ligada a otros agentes virales como el citomegalovirus (CMV) y el virus de la hepatitis C (VHC).3 La evaluación retrospectiva del registro estadounidense Scientific Registry of Transplant Data, documentó los datos de 210 763 pacientes postrasplante de órgano sólido (TOS), de los cuales 1630 presentaron PTLD. La distribución de los casos mostró dos picos de incidencia tras la intervención, al año (3.2 por 1000 personas/año), y a los nueve años (3.1 por 1000 personas/ año). La infección crónica por el VHC no se asoció en esta cohorte a un riesgo aumentado de PTLD, en especial tras ajustar los valores según el tipo e indicación del trasplante, por el grado de incompatibilidad del HLA, el tipo de donante y la medicación inmunosupresora.8
El EBV fue el primer virus relacionado con el desarrollo de PTLD, condición que presenta un amplio espectro nosológico, oscilando desde las lesiones indolentes con patrón policlonal que resuelven usualmente con la reducción de la inmunosupresión, hasta los linfomas agresivos de evolución rápida que con frecuencia no responden al tratamiento citotóxico. La clasificación de Harris, adoptada por la Organización Mundial de la Salud (OMS), divide los PTLD en cuatro grupos: lesiones tempranas, polimórficas, monomórficas, y aquellas similares al linfoma Hodgkin de patrón clásico.9 Las decisiones terapéuticas se basan con frecuencia en el subtipo histológico, grado tumoral, estado funcional del paciente, sitio de origen y estado del órgano trasplantado, que al menos en parte, limita la tolerancia al tratamiento oncológico.10
La mayoría de los pacientes con PTLD presentan una carga viral positiva para EBV, un virus herpes gamma identificado inicialmente en el cultivo de células del linfoma Burkitt.11 La fisiopatología derivada de la inmunosupresión terapéutica en los pacientes receptores de TOS explica el riesgo de aparición relacionada con la disminución de los linfocitos T citotóxicos y con la seronegatividad para el virus antes del trasplante.3 El EBV infecta las células B interactuando con el CD21 un componente primario del receptor para el complemento (fracción C3b inactiva), por el cual se endocita el genoma viral que es transportado e incluido al núcleo, donde favorece la reconfiguración de la información genética del hospedero, antes del inicio de la replicación.11 Múltiples estudios han demostrado que cerca de 1% de las células B infectadas entran en autolisis promoviendo la multiplicación viral y favoreciendo la expresión de genes agrupados en un formato latente (en contraposición a los líticos) que mantienen la infección, incluyendo los antígenos nucleares del EBV (EBNA 1 a 6) y las proteínas latentes de membrana (LMP1, LMP2A y LMP2B). Estos genes se pueden caracterizar en cuatro programas diferenciaes de expresión (latencias tipo 0, I, II y III), entre los cuales se encuentran el de crecimiento celular y expansión clonal, que conlleva a la pérdida de los mecanismos de control de la proliferación.12,13 La biología del EBV se suma a múltiples factores de riesgo propios del huésped, eventos que explican la génesis del PTLD. La Tabla 1 agrupa los factores de riesgo relacionados de forma significativa con el desarrollo de la enfermedad.
No existe consenso respecto del tratamiento óptimo para los pacientes con PTLD y pocos estudios prospectivos no aleatorizados con limitado tamaño de muestra han evaluado la eficacia y seguridad de diversos medicamentos. Se ha intentado aplicar cirugía, radioterapia, inmunoterapia (interferón), quimioterapia y agentes biológicos, con una morbilidad y mortalidad que oscilan entre 30% y 50%.14 Las aproximaciones más recientes postulan el uso de agentes antivirales y linfocitos T citotóxicos HLA compatibles y específicos estimulados contra el EBV.12 Recientemente, el rituximab (Mabthera®), un anticuerpo monoclonal humanizado anti-CD20, utilizado para el tratamiento del linfoma folicular y de la neoplasia B difusa de células grandes, ha sido utilizado con éxito en pacientes con PTLD.15,16 Los resultados de algunos estudios retrospectivos parecen prometedores, pero resultan inconsistentes, debido a la divergencia en las tasas de respuesta que se encuentran entre 20% y 100%. Con el fin de resolver dicha heterogeneidad, Choquet y colaboradores desarrollaron un experimento clínico fase II, que incluyó 46 pacientes con PTLD de estirpe B (hasta ahora es el estudio publicado de mayor dimensión) diagnosticado después del TOS y tratado con rituximab como monoterapia. Se evidenció una tasa de respuesta completa de 44%, supervivencia global a un año de 67% y tasa de control de la enfermedad en este término de 68%. El único factor que predijo la respuesta después de 80 días del tratamiento, fue la normalización en los niveles iniciales de la lactato deshidrogenasa (OR = 6.9; p = 0.07).17
A continuación se presenta el caso de un paciente diagnosticado con PTLD hepático CD20 negativo post-trasplante, tratado con rituximab, medicamento con el cual alcanzó remisión completa. Los autores exploran algunas hipótesis respecto del sustento biológico para este hallazgo y realizan un recuento detallado de las opciones terapéuticas.
¿ PRESENTACIÓN DEL CASO
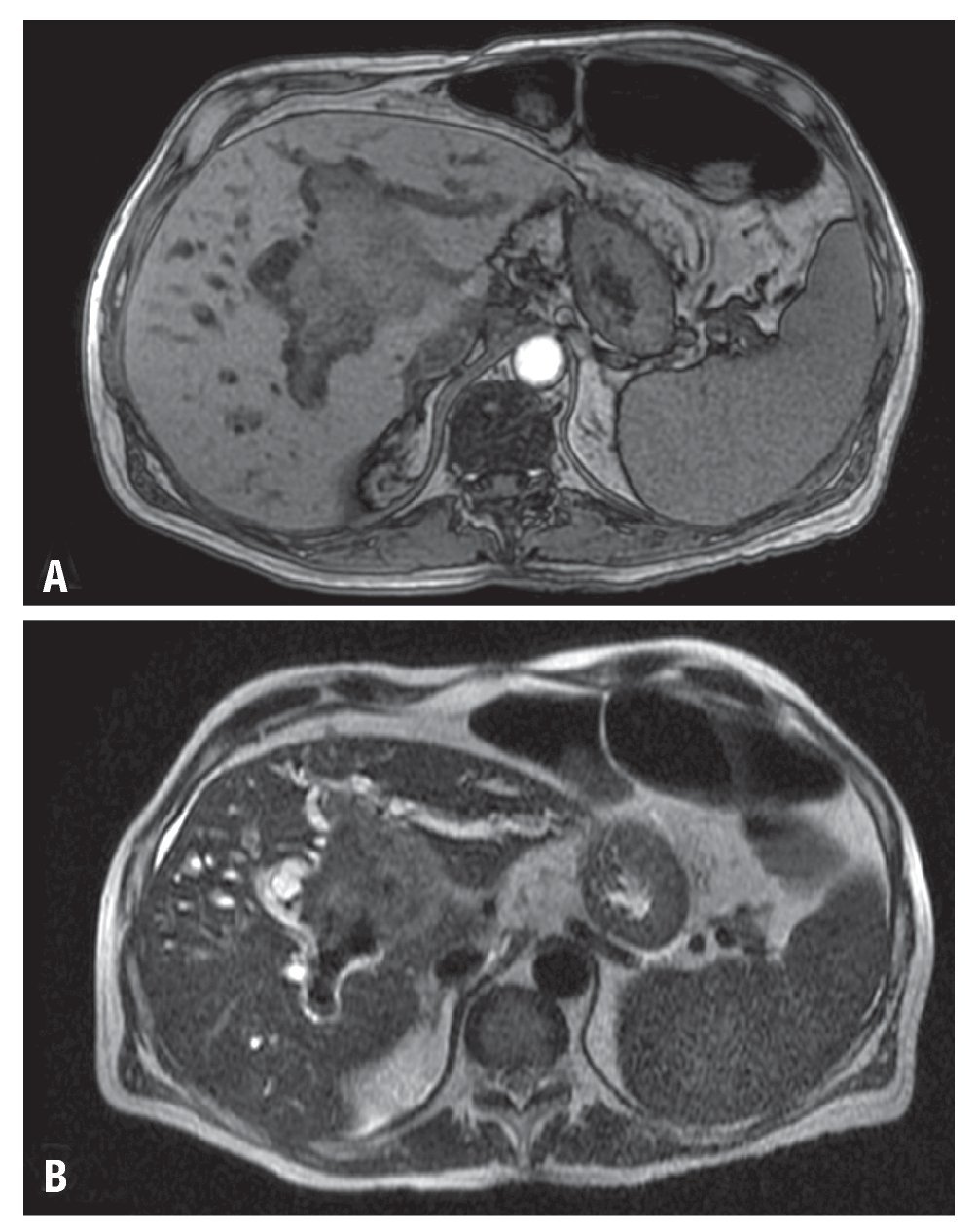
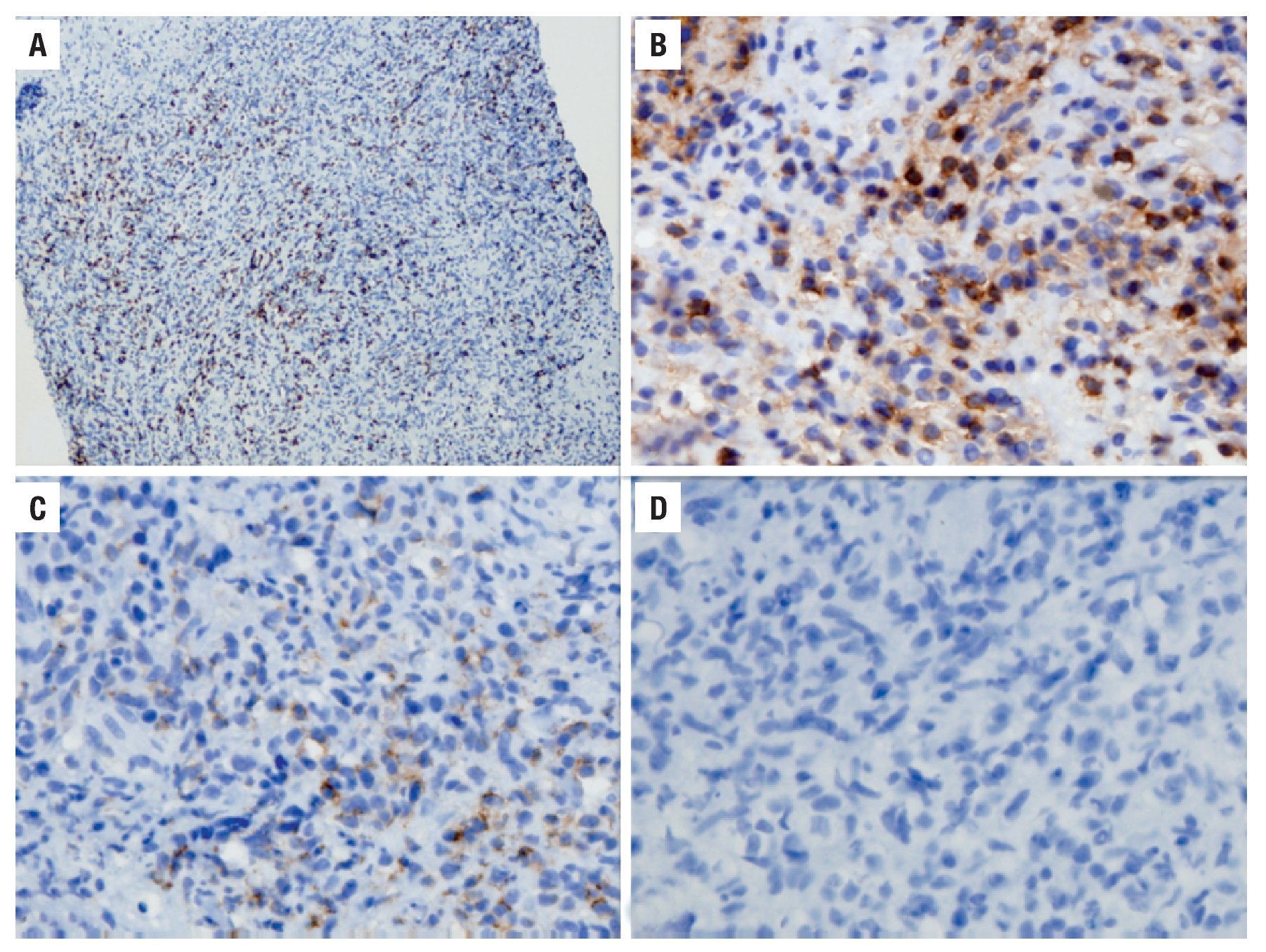
Hombre de 68 años, profesor universitario con diagnóstico pre-trasplante de cirrosis hepática por hemocromatosis desde 1997 controlada con flebotomías y manejo médico. Al diagnóstico también se documentó un rasgo talasémico sin estudios genéticos complementarios. Durante 2007 se demostró aumento progresivo del perímetro abdominal por ascitis, acompañada de encefalopatía y deterioro de la función hepática, con diagnóstico de cirrosis Child-Pugh C (puntaje de 10) y puntuación por la escala de MELD de 17. Como antecedentes presentaba diabetes mellitus insulinodependiente con insuficiencia renal crónica estadio tres, hipotiroidismo y polineuropatía multifactorial de predominio sensitivo en los miembros inferiores. En junio de 2008 se le realizó un trasplante de hígado ABO relacionado, sin complicaciones. Posteriormente, inició inmunosupresión triple convencional con ciclosporina, micofenolato y prednisolona; fue dado de alta sin complicaciones al quinto día posoperatorio. En consulta externa, se cambió el inhibidor de calcioneurina de ciclosporina a tacrolimus (primer mes) y posteriormente de éste a sirolimus (segundo mes) por cuadro de hiperkalemia secundaria a inhibidores de calcioneurina. En junio de 2009 se evidenció ictericia progresiva en relación a la aparición de una lesión hepática en resonancia magnética nuclear con densidad de tejidos blandos que ocasionaba compresión de la vía biliar (Figura 1). La serología para EBV fue negativa para IgM y positiva para IgG; no se realizó cuantificación de la carga viral. En ese momento, se practicó colangiografía pancreática retrograda endoscópica (CPRE) con papilotomía y colocación de stent. Se realizó entonces una biopsia hepática, cuyo estudio mostró infiltración difusa por una población heterogénea de células de aspecto plasmocitoide entremezcladas con células grandes atípicas acompañadas de necrosis; las células neoplásicas mostraron reactividad exclusiva para CD45 y MUM1, sin evidencia de expresión de marcadores B tales como CD20, CD79A, y PAX5, ni del CD3. La actividad proliferativa cuantificada con el Ki67 fue cercana al 50%. Con estos hallazgos se hizo el diagnóstico de un linfoma difuso de célula grande de linaje no definido compatible con un PTLD. Se documentó además en una subpoblación celular la expresión de la proteína tardía de membrana del EBV (LMP1) (Figura 2).
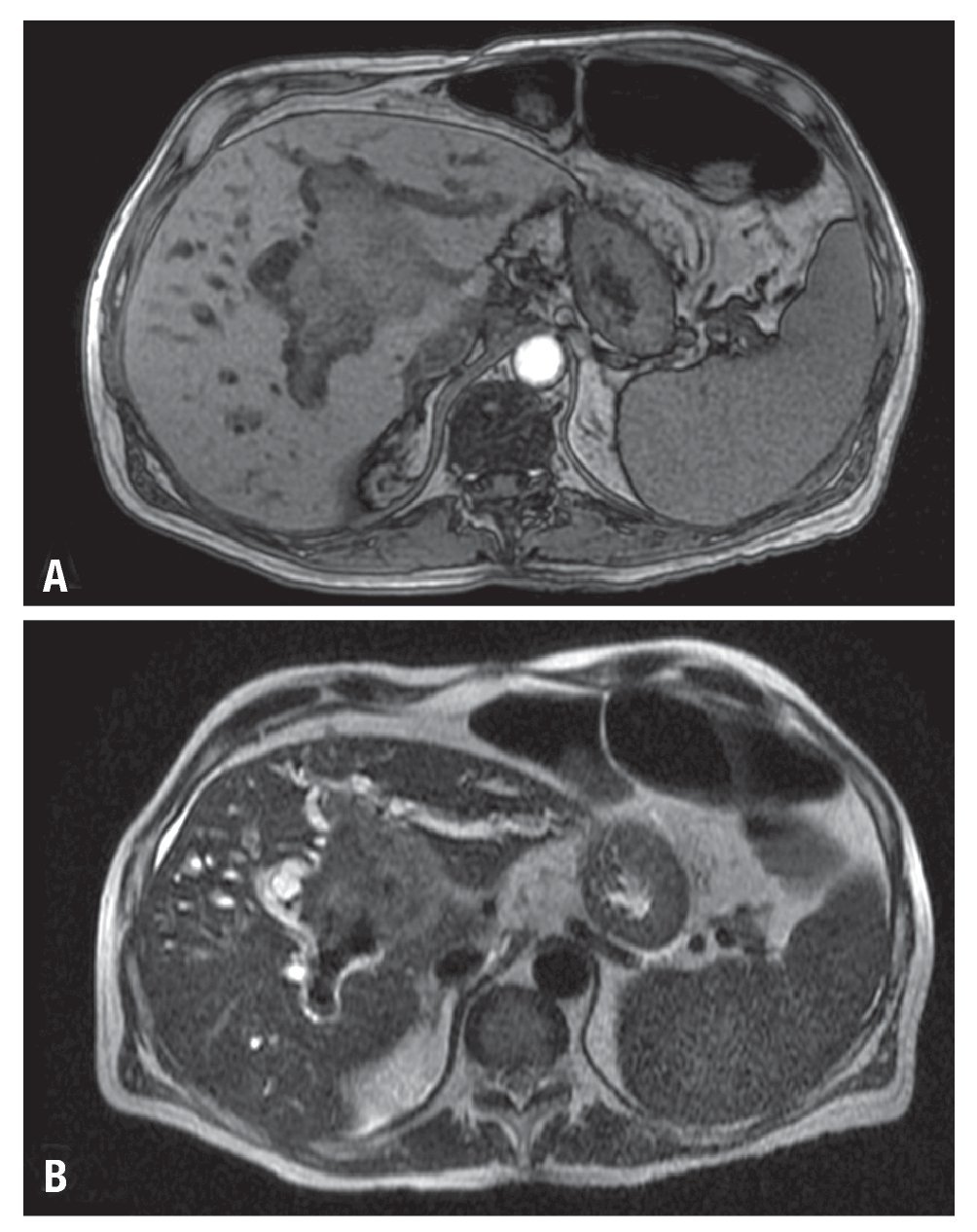
Figura 1. Imágenes de la resonancia magnética del hígado. Secuencias con información T1 (A) y T2 (B), que demuestran lesión neoplásica en aspecto central denle el lóbulo izquierdo, de 7 cm de diámetro mayor, condicionando obstrucción de vía biliar, con dilatación intra-hepática.
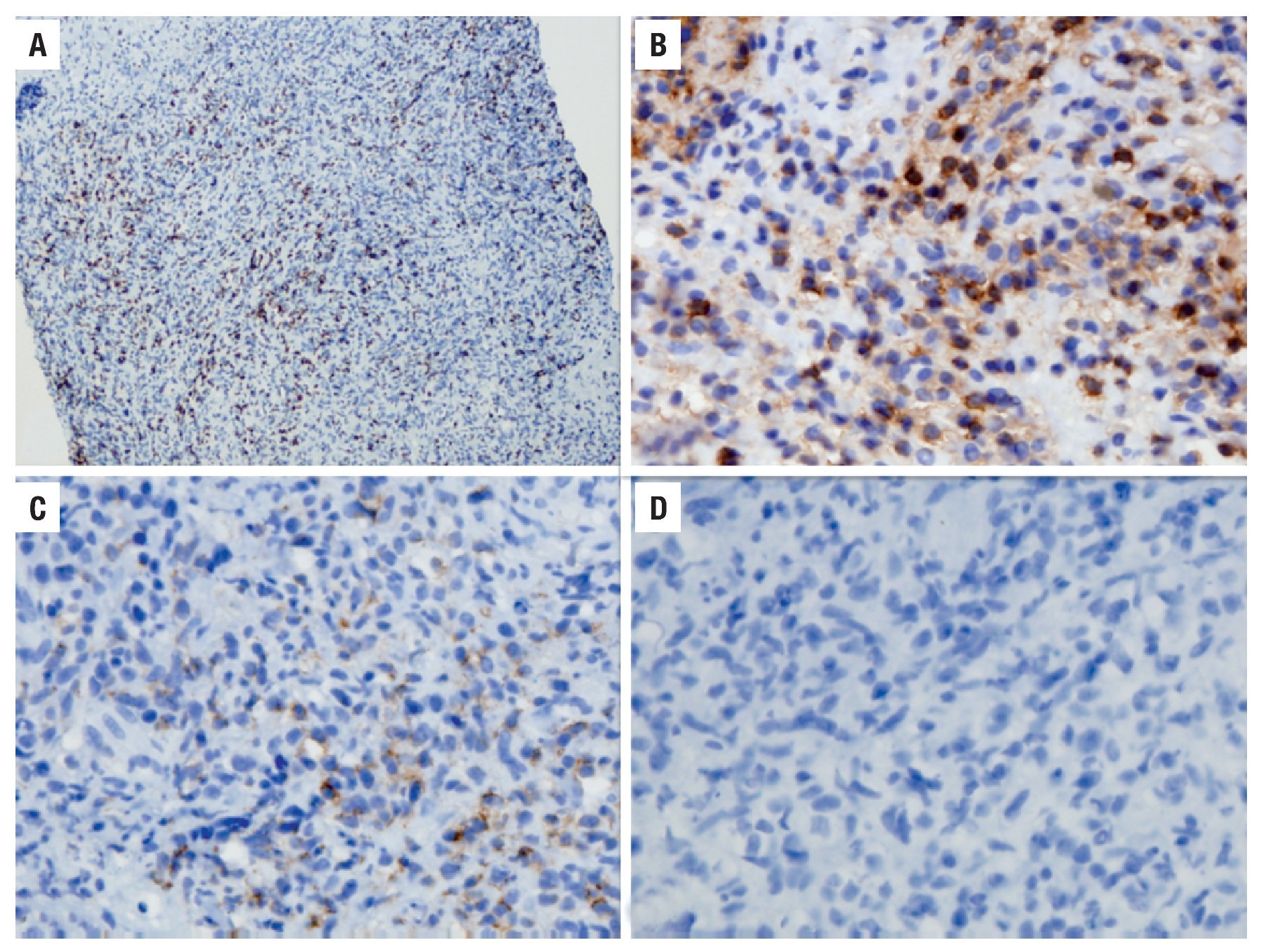
Figura 2. Estudio histopatológico de la lesión. A. Estudio de inmunohistoquímica con positividad para el marcador KI67, que demuestra una actividad proliferativa de 50% (10 aumentos). B. Estudio de inmunohistoquímica con positividad para el marcador CD45, confirmando el origen linfoide de la lesión (20 aumentos). C. Estudio de inmunohistoquímica con positividad focal para el marcador LMP1, que demuestra infección activa por EBV (40 aumentos). D. Estudio de inmunohistoquímica con negatividad para el marcador CD20 (40 aumentos).
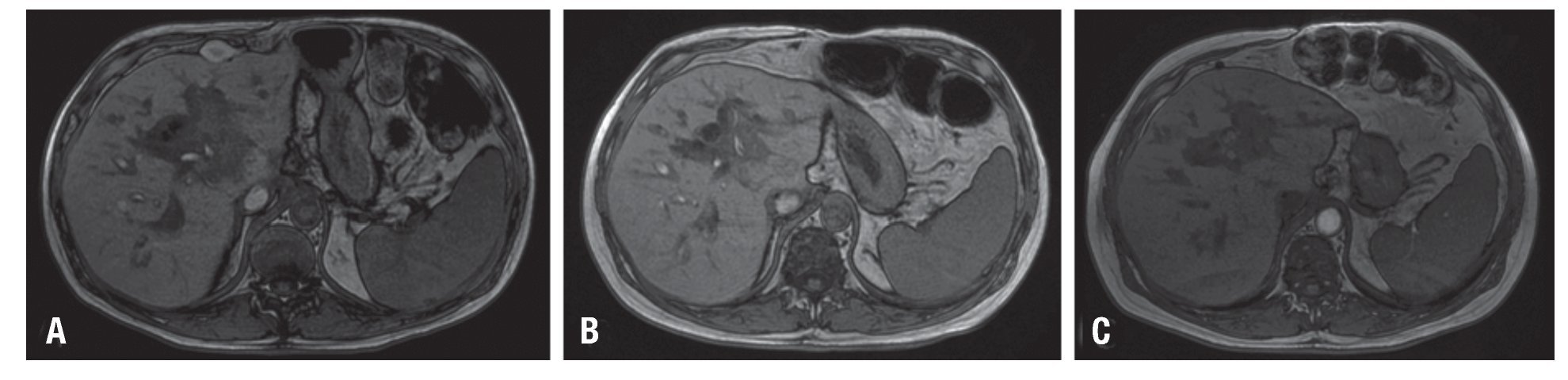
Se inició manejo mediante la disminución de la inmunosupresión con sirolimus en 40% y prednisona en 50% experimentado empeoramiento clínico y aumento del patrón colestásico y por lo que tras dos semanas se inició tratamiento con rituximab 375 mg/m2 semanal por cuatro semanas, presentando tempranamente bacteremia por Escherichia coli interpretada como posible colangitis pos-CPRE manejado con ertapenem. El seguimiento se realizó con resonancia magnética nuclear dos meses después de finalizada la terapia, encontrando una respuesta cercana a 40% de acuerdo a la reducción del volumen tumoral por criterios de Cheson (Figura 3A). Dicho hallazgo se consideró aceptable, debido al esquema utilizado, y a la mejoría del paciente, por lo que se decidió continuar con un segundo ciclo de rituximab en agosto de 2009 con buena tolerancia. La respuesta evidenciada por imágenes tras dos ciclos fue de 80% (Figura 3B). Concomitantemente se realizó seguimiento del recuento de células T CD4+ encontrando un nivel de 154 células/mL, motivo por el cual se administró profilaxis con trimetoprim/sulfa-metoxazol y fluconazol, por lo que se suspendió el monoclonal. Luego de completar tres meses sin rituximab, se encontró recuperación de los linfocitos T CD4+, retirando la profilaxis y para seguir con conducta expectante. La evaluación de seguimiento practicada en enero de 2010 encontró una remisión completa persistente (Figura 3C); veintiún meses después del diagnóstico de PTLD, el paciente se encuentra asintomático, en seguimiento y libre de enfermedad con injerto viable y funcional.
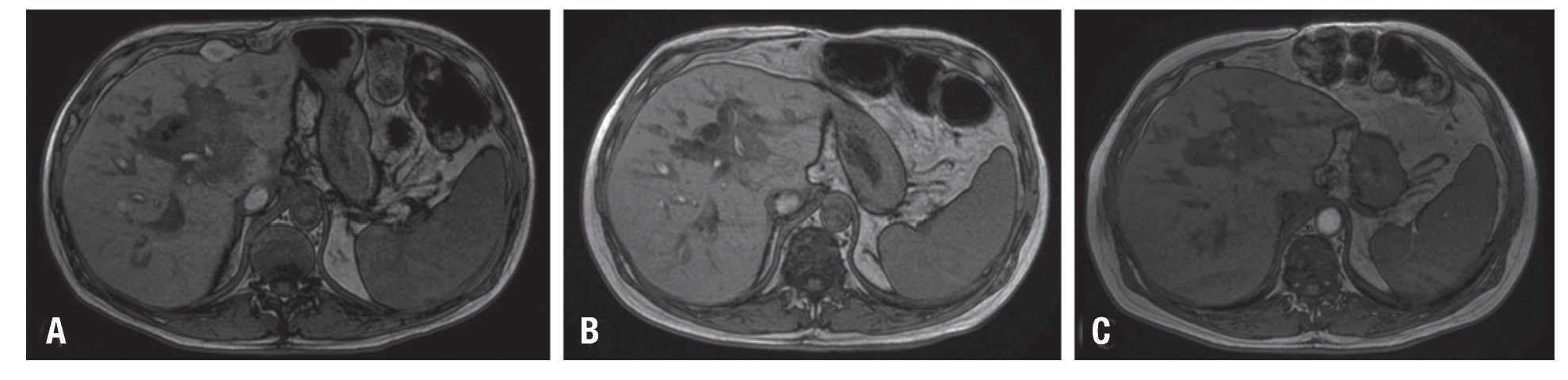
Figura 3. Imágenes de resonancia magnética del hígado. Seguimiento por imágenes sobre la respuesta al tratamiento con información T1, transcurrido un mes tras un ciclo de rituximab (A), transcurridos tres meses tras dos ciclos de rituximab (B) y luego del primer año de seguimiento (C). Las imágenes muestran disminución significativa del tamaño de la lesión descrita, inicialmente determinada por disminución del diámetro mayor de 40% (A), posteriormente de 80% (B) y en remisión completa sin aplicación de nuevas dosis de rituximab (C). El efecto obstructivo sobre la vía biliar persiste, con discreta dilatación de vía intra-hepática.
¿ DISCUSIÓN
La presentación clínica de los pacientes con PTLD es variable e incluye fiebre, linfadenomegalias, pérdida de peso, perforación intestinal y sepsis. Con frecuencia, se evidencia compromiso extraganglionar en el hígado, pulmón, intestino, riñón, médula ósea y en la piel.12 En una cohorte que incluyó 7040 pacientes receptores de TOS tratados en la Universidad de Michigan se encontraron 78 casos de PTLD tras 43 años de seguimiento; en general, los pacientes presentaban una edad promedio de 40 años, 27% tenía síntomas B asociados, y 78% presentó compromiso tumoral extraganglionar. El subtipo más frecuente de PTLD en estos casos fue el Linfoma B Difuso de Célula Grande (LBDCG) documentado en 55%. El registro de seguimiento encontró una mediana para la supervivencia global de 8.2 años.18
La presencia de colestasis como manifestación inicial del PTLD, es infrecuente; al igual que en nuestro caso, Baron y colaboradores describieron una serie de 14 pacientes que debutaron con una masa localizada en la porta-hepatis, que generó compresión extrínseca de la vía biliar y colestasis en pacientes con trasplante hepático. Dicho estudio documentó un tiempo promedio de cinco meses entre la realización del TOS y el diagnóstico de PTLD, tiempo que en nuestro paciente fue de un año. No todos los pacientes fueron seguidos a largo plazo, pero algunos alcanzaron una supervivencia superior a los 24 meses. Los autores alertaron sobre la necesidad de considerar al PTLD localizado a nivel hepático como una alternativa diagnóstica en los casos de ictericia obstructiva documentada tres a seis meses después del TOS.19
El análisis genómico de las inmunoglobulinas en las células del PTLD, ha demostrado que la gran mayoría de estas lesiones derivan del centro germinal y de las células localizadas en la región pos-germinal del ganglio linfático. Aproximadamente, 50% de las células tumorales presenta una pérdida funcional del receptor B.20 La proliferación de las células B, observada en personas sanas que presentan la infección por EBV suele ser policlonal, mientras las alteraciones asociadas con el PTLD, pueden ser policlonales, oligoclonales o monoclonales.21 La progresión de una lesión policlonal con extenso compromiso visceral se asocia a la activación de la capacidad oncogénica de los factores virales que promueven la activación del factor nuclear κB (NFκB), sobrerregulación del gen MYC y represión del complejo mayor de histocompatibilidad clase I.11 Adicionalmente, las anormalidades citogenéticas parecen raras en el PTLD, a pesar de que los linfocitos B en fase de proliferación, pueden adquirir una serie de cambios que incluyen la inestabilidad micro-satelital, defectos en la capacidad de reparación del ADN, mutaciones en el proto-oncogen MYC, y en los genes BCL6 y P53. También se ha descrito hipermetilación y mutaciones somáticas aberrantes hasta en 30% de los PTLD monomórficos, particularmente, en aquellos que siguen una distribución tipo B.22,23
El PTLD de estirpe B asociado a negatividad para el receptor CD20, puede explicarse por la reprogramación celular debido a un fenómeno de regulación hacia abajo de los marcadores B secundario al efecto inhibitorio de la proteína latente de membrana 2 (LMP-2). Además, se presume que debido a la ausencia de CD20, la inmunoterapia específica con rituximab puede ser inefectiva en estos casos.11 Por otro lado, en los sujetos tratados con rituximab que luego presentan resistencia, se presume un efecto de selección clonal asociada a subexpresión del receptor CD20. Un estudio previo encontró 12% de LBDCG CD20 negativo, manifestación que se asoció con una menor supervivencia global versus la contraparte CD20 positivo (HR 0.20, IC95% 0.06-0.66; p = 0.01).18 Por el contrario, en nuestro caso a pesar de la negatividad para CD20 desde el inicio de la enfermedad, la terapia con rituximab fue efectiva y sostenida.
El mecanismo de acción principal del rituximab es la inducción de la muerte en células tumorales CD20 positivo, siguiendo la citotoxicidad directa mediada por el complemento, a través de la inducción de apoptosis, o sencillamente, por disminución del metabolismo celular.24
La inmunosupresión necesaria para mantener la viabilidad del injerto en todo paciente trasplantado está dirigida a bloquear la acción citotóxica propia del linfocito T CD8+, intervención que favorece la proliferación de las células B infectadas. Desde el punto de vista histopatológico parece claro que la morfología del PTLD incluye en la periferia un halo conformado por células T CD4+, encargadas de proteger los elementos tumorales, aumentando la proliferación e inhibiendo la apoptosis.25 La regresión en el recuento de células T CD4+ en nuestro paciente podría estar en relación a una disminución de la presentación de antígenos, evento descrito en modelos animales y humanos de enfermedades autoinmunes.26 Además, este fenómeno se ha asociado a la disminución en la producción de citoquinas proinflamatorias por las células B como interleucinas 1, 10, 6, interferón-γ (IFN-γ) y factor de necrosis tumoral α (TNF-α).
La terapia con rituximab puede producir inmunodepresión por estos mecanismos, además del efecto descrito sobre células B, evento que podría limitar su uso por complicaciones infecciosas. No obstante, el monoclonal, también es capaz de ejercer cambios en la población T, por el estímulo negativo de las células presentadoras de antígenos CD20+.27 Por otro lado, el tratamiento ideal del PTLD debería incluir la administración de agentes que permitan la reconstitución de la actividad de las células T CD8+ específicas contra el EBV. Este objetivo puede lograrse, entre otros, con la reinfusión de linfocitos T CD8+ EBV específicos y HLA compatibles, intervención que incrementa el riesgo de activar la enfermedad injerto contra huésped, resulta extremadamente costosa y difícil de establecer por la disponibilidad de donantes, sólo es aplicable a los casos EBV positivos y por el momento se recomiendan usar sólo en el contexto de experimentos clínicos.12,28
Recientemente, el British Committee for Standards in Haematology (http://www.bcshguidelines.com/) publicó unas guías dirigidas a difundir información sobre el control del PTLD. Dentro de las recomendaciones, se sugirió reducir la inmunosupresión (grado B, nivel 3 de evidencia), y el inicio de rituximab como agente único administrado a los sujetos que no tienen respuesta después de una reducción programada y paulatina de la inmunosupresión (grado B, nivel 3 de evidencia).28
Al menos un estudio valoró la eficacia y seguridad del esquema R-CHOP, que se reserva para los sujetos con PTLD con compromiso orgánico crítico o con diagnóstico de linfoma clínicamente agresivo (grado C, nivel 4 de evidencia), que no logren entrar en remisión completa o que progresan a pesar de la reducción de las dosis de los inmunosupresores y de la administración del monoclonal como monoterapia, en parte debido a la toxicidad asociada.28 En nuestro caso, se disminuyó la dosis del sirolimus sin mejoría evidente, motivo por el cual, se administró rituximab logrando remisión completa después de ocho semanas de tratamiento secuencial, sin requerimiento de quimioterapia concomitante.
Aunque en el presente caso, la respuesta a la inmunoterapia fue excelente a pesar la negatividad para el CD20, la evidencia proveniente de estudios retrospectivos sugiere que su efectividad puede estar ligada a otros mecanismos de acción adicionales al de este receptor de membrana, ya que su negatividad no contraindica el uso en los PTLD de estirpe B. Estos casos deberían ser seguidos prospectivamente de manera cautelosa con el fin de evidenciar tempranamente la recaída, que puede ser refractaria al tratamiento único con inmunoterapia. Los autores consideran posible que el efecto del rituximab además comprenda la inmunomodulación de las células T dispuestas en la periferia tumoral.
Hay controversia respecto a la utilidad de la determinación de la carga viral del EBV en el contexto del PLTD. En primer lugar, se postuló que la carga viral podría tener una repercusión como ayuda diagnóstica en casos dudosos y relacionarse con la respuesta tumoral al tratamiento en los casos EBV+, a pesar de que no constituye una prueba específica y que el estándar de oro es el estudio histopatológico.25 Se ha estimado que una medida de ADN del EBV libre mayor de 10.000 copias/ mL en el contexto de pacientes pos-TOS se correlaciona con el diagnóstico de PTLD en 100% de los casos.15 Durante el tratamiento, la disminución del volumen tumoral puede asociarse inicialmente con un aumento del nivel circulante del ADN viral por liberación de los antígenos presentes en las células tumorales fraccionadas, y posteriormente puede disminuir como consecuencia de la eliminación de los elementos infectados.11 A pesar del pobre nivel de evidencia la American Society for Transplantation (AST; http://www.a-s-t.org/) recomienda la cuantificación de la carga viral al momento del diagnóstico del PLTD en todo paciente con TOS.29 En segundo lugar, evaluar la carga viral podría ser útil para determinar tempranamente el riesgo de TOS en pacientes con factores adversos, en quienes podría ser útil el inicio de profilaxis (por ejemplo, en la población pediátrica menor de un año y en los receptores seronegativos para EBV al momento del trasplante). De acuerdo con la AST, se recomienda la determinación mensual de la carga viral durante el primer año postrasplante, y tras este periodo realizar un seguimiento regular en sujetos con carga viral elevada persistente pero estable, y en aquellos que aún reciben inmunosupresión intensiva. El monitoreo selectivo se recomienda en los casos seropositivos, en especial para niños y en aquellos en quienes se cambia el esquema inmunosupresor.29
Otra estrategia terapéutica recientemente descrita, postula el uso de medicamentos antivirales (ganciclovir, aciclovir, valaciclovir) como manejo coadyuvante del PTLD, en particular en los casos EBV+ con lesiones tempranas y neoplasias polimórficas.28 La eficacia terapéutica de esta opción se encuentra cuestionada, como parte del manejo de las lesiones malignas. Fisiopatológicamente, la utilidad de los antivirales se explica debido a que las células transformadas no expresan la enzima viral timidina quinasa, necesario para la fosforilación de los análogos de la guanosina, lo que la transforma en una sustancia bioactiva capaz de inhibir su incorporación por la ADN polimerasa viral.25 Otra opción a considerar, es el uso de la arginina butirato concomitantemente con los antivirales; este compuesto ha demostrado in vitro la inducción de la expresión de esta enzima, convirtiendo a las células infectadas susceptibles a la acción de medicamentos dirigidos contra el EBV.12 Actualmente, hay tres experimentos clínicos registrados en ClinicalTrials. gov diseñados para evaluar el uso de este agente en los casos de neoplasias linfoides EBV+, asociándolo a la terapia antiviral.
¿ CONCLUSIONES
Se presentó el caso de un paciente postrasplante hepático que luego del primer año presentó manifestaciones clínicas secundarias a la presencia de una masa, en parénquima del órgano trasplantado con efecto obstructivo sobre la vía biliar, cuya histología demostró un PTLD que resultó histológicamente negativo para CD20. El paciente recibió tratamiento con disminución de la inmunosupresión y rituximab, logrando remisión completa y sostenida hasta la fecha. La negatividad para el receptor CD20 no contraindicó, ni limitó el uso de este tipo de inmunoterapia en un paciente con PTLD. Se presume que la utilidad del rituximab estuvo asociada con la disminución del recuento de linfocitos T CD4+.
¿ AGRADECIMIENTOS
Los autores desean agradecer la colaboración del Servicio de Anatomía Patológica, en especial a la Dra. Rocío López y el Dr. Rafael Andrade. Adicionalmente al Servicio de Imágenes Diagnósticas del Hospital Universitario Fundación Santa Fe de Bogotá, en especial al Dr. Diego Aguirre, por su colaboración en la preparación del caso clínico.
Correspondencia: Dra. Myriam Rodríguez.
Médico Internista y Hematólogo. Calle 119, N°7 - 75, Bogotá, Colombia.
Teléfono: 571 603 0303, ext. 5227. Fax: 571 657 5714.
Correo electrónico: ml.rodriguez77@uniandes.edu.co