


El cáncer rompe las reglas básicas del comportamiento celular, mediante el cual los organismos multicelulares están regidos. Los productos de los oncogenes y genes supresores tumorales modifican diferentes vías de señalamientos intracelulares fisiológicas, alterando los programas de proliferación, diferenciación apoptosis, adhesión/movilidad, entre otros. En el cáncer como en otras complejas enfermedades, las alteraciones de las vías de señalamientos intracelulares de las células transformadas, se encuentran bajo el efecto de células del microambiente tisular o células localizadas a distancia, a través de señalamientos extracelulares. Los diferentes tipos de cáncer no corresponden a una sucesión fija de mutaciones específicas de varios genes, por el contrario diferentes productos de la expresión de genes son modificados por alteraciones genéticas, epigenéticas progresivas. Estas moléculas modificadas participan en diferentes rutas como componentes de redes moleculares de las vías intracelulares de señalamientos fisiológicas, y consecuentemente las transforman en vías de señalamiento oncogénicas, que finalmente conducen a la iniciación y progresión del cáncer, incluso en cánceres clínicamente idénticos. En esta revisión se describen los diferentes conceptos clásicos y modernos, que permiten aumentar el entendimiento de la iniciación y progresión del cáncer como un sistema biológico, entre ellos: oncogenes, genes supresores tumorales, genes conductores y pasajeros del proceso, las principales modificaciones genómicas, epigenómicas y proteómicas, las vías de señalamientos oncogénicos, los fenotipos oncogénicas y las relaciones entre las células tumorales y las células de su microambiente tisular.
Cancer cells take no notice of basic environment human tissues. The products of oncogenes and tumor suppressor genes expression alter the physiological intracellular signaling pathways, and deregulating proliferation, differentiation, apoptosis, adhesion/mobility cell programs, etc. In cancer as other complex diseases, the intracellular signaling pathways modifications, are under the influence of several cells of the microenvironment tissue or located from a distance through extracellular signaling. Different cancer types are not a strictly deterministic gen-mutations disease; rather these genes products modified by genetic/epigenetic alterations. Then, these modify molecules participate in different routes as net-molecularly component of the physiological intracellular signaling pathways and transform it in oncogenic signaling pathways, and finally drive to the cancer initiation and progression, even clinically identical cancers. In this review, we describe the classical and the modern concepts to let a improvement of the cancer initiation and progression understanding as a biological system, as: oncogenes, tumor suppressor genes, driver and passenger genes, genomic, epigenomic and proteomic alterations, oncogenic signaling pathways, oncogenic phenotype, and tumor cells and microenvironment tissue cells interactions.
¿ INTRODUCCIÓN
Las células que constituyen un organismo eucarionte complejo funcionan como una sociedad o un ecosistema, se reproducen por división celular y se organizan en colaboración formando tejidos. La organización celular esta jerarquizada para mantener su supervivencia, de tal forma que las células germinales al diferenciarse en células somáticas preserven copias de sus propios genes. Para coordinar su comportamiento, las células mandan, reciben e interpretan una serie de señales extracelulares, que son órdenes de cómo deben comportarse en cuanto a permanecer latentes, crecer, dividirse, diferenciarse o morir, de acuerdo a las necesidades del organismo.1
El cáncer rompe las reglas básicas del comportamiento celular, mediante el cual los organismos multicelulares están regidos. Las células tumorales presentan defectos en los circuitos regulatorios que controlan la proliferación celular normal y su homeostasis. Las células cancerosas presentan dos principales características hereditarias: se reproducen sin ajustarse a las restricciones normales del crecimiento y división celular, e invaden y colonizan territorios normalmente destinados a otras células. Un tumor deriva de la división de una sola célula transformada por daño génico, el cual acumula y transmite a algunos de sus descendientes; el análisis molecular de los cromosomas en las células tumorales ha demostrado su origen clonal (por ejemplo, en leucemia mielocítica crónica, por la identificación del cromosoma filadelfia).2
El advenimiento de secuenciación genómica ha permitido el entendimiento del cáncer en términos de mutaciones de oncogenes y genes supresores tumorales. Los estudios de interferencia del RNA, han mostrado que la desregulación de diferentes productos génicos, contribuye al fenotipo tumoral celular. El producto de la expresión de un gen, puede identificarse como un componente de una vía de señalamientos y/o en la interacción con múltiples vías de señalamientos intracelulares de regulación transcripcional, metabólicas, programación funcional, entre otros. Asimismo, en el cáncer como otras complejas enfermedades, las alteraciones de las vías de señalamientos intracelulares de las células transformadas, se encuentran bajo el efecto de células del microambiente tisular o células localizadas a distancia, a través de señalamientos extracelulares. Los productos de los oncogenes y genes supresores tumorales modifican diferentes vías de señalamientos intracelulares fisiológicas, alterando los programas de proliferación, diferenciación y apoptosis, adhesión/movilidad, entre los más importantes.3
Actualmente, es evidente que el cáncer no es una enfermedad determinística, que solo depende de la sucesión fija de mutaciones específicas de varios genes. Al contrario, estos productos participan en diferentes rutas de las vías de señalamientos intracelulares fisiológicas, los cuales las convierten en vías oncogénicas que conducen a la iniciación y progresión del cáncer, incluso en cánceres clínicamente idénticos. El desarrollo final de un cáncer en un ser humano, está influenciado por otros múltiples factores que comprenden la respuesta inmunológica, la edad, la nutrición y el microambiente tisular. Los tumores pueden considerarse como una colección, que acumula alteraciones genéticas y epigenéticas, las cuales son influenciadas por las diferentes células vecinas, siendo el flujo de esta interacción en ambos sentidos. La inestabilidad genómica es una característica fundamental de las células cancerosas, sin embargo diferentes factores extrínsecos no-genéticos (epigenéticos), participan significativamente y agregan mayor complejidad al inicio, y a la progresión de la transformación maligna.3,4
¿ ALTERACIONES GENÉTICAS Y EPIGENÉTICAS
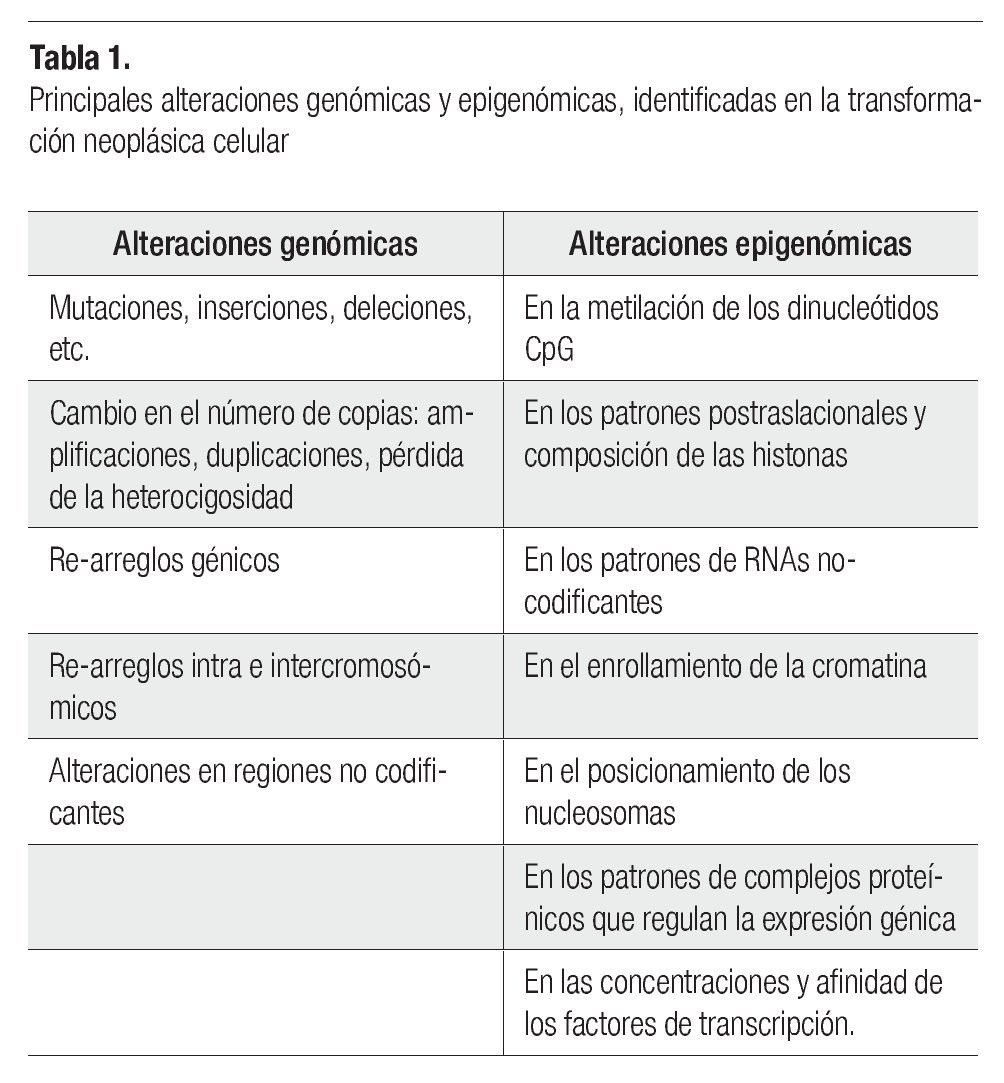
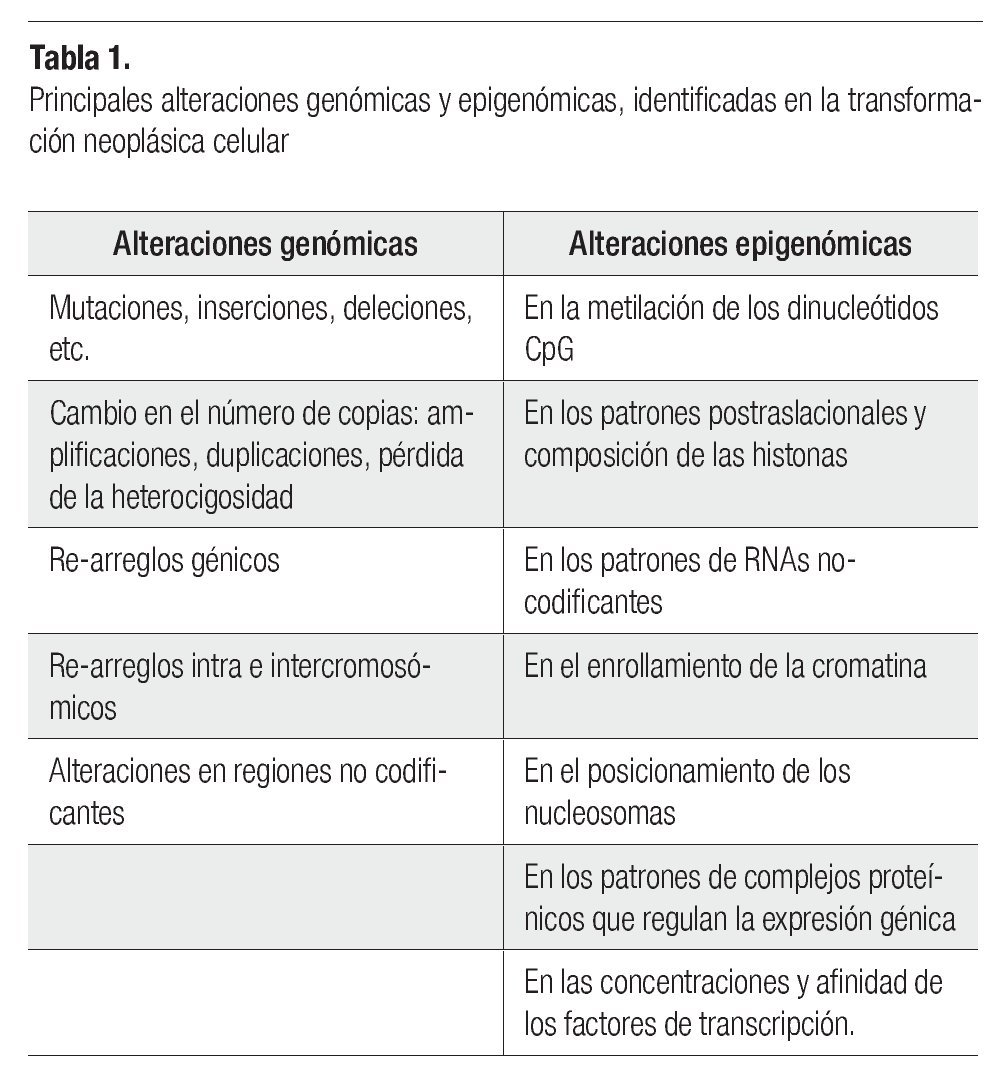
El desarrollo del cáncer en los individuos adultos depende de mutaciones somáticas, favorecidas en las personas quienes tienen defectos hereditarios en los genes de uno o algunos de los sistemas de reparación del DNA, o eventualmente presentan polimorfismos genéticos (no protectores) de enzimas involucradas en el metabolismo de sustancias o agentes carcinogénicos, provocando que sus células acumulen tasas elevadas de mutaciones. El desarrollo de los tumores avanza por un proceso análogo a la evolución darwiniana, en el cual la sucesión de cada cambio genético le confiere al siguiente, una ventaja en el crecimiento celular que conduce a la transformación progresiva de células normales a células cancerosas.5 Uno de los problemas para entender el cáncer es identificar si las alteraciones genómicas en las células transformadas, son debidas a cambios genéticos (en la secuencia del DNA) o a cambios epigenéticos (cambios persistentes en la expresión génica, sin cambios en la secuencia del DNA, por medio de modificaciones en las histonas de los nucleosomas y en la metilación del DNA) (Tabla 1). La carcinogénesis está ligada a la mutagénesis (producción de un cambio en la secuencia del DNA), lo cual es evidente para los carcinógenos químicos (que provocan cambios en la secuencia de los nucleótidos), y para las radiaciones, como los rayos-X (que causan ruptura de los cromosomas, y translocaciones).
Una sola mutación no es suficiente para causar cáncer, un gran número de evidencias experimentales y estudios epidemiológicos, indican que el desarrollo del cáncer requiere de varias alteraciones genéticas.6,7 Si una mutación fuera suficiente para desarrollar cáncer, la incidencia de los pacientes con cánceres no-hereditarios sería igual en cualquier edad, sin embargo como se ha demostrado la incidencia se relaciona directamente con la edad. Muchas de las mutaciones específicas relacionadas con el desarrollo del cáncer, han sido identificadas gradualmente en periodos de varios meses y años posteriores a la exposición, en individuos expuestos a agentes carcinogénicos. Esta información ha permitido al mismo tiempo, establecer diferentes estrategias de prevención primaria o secundaria en poblaciones con riesgos elevados, al desarrollo de algunos tipos de cáncer. Una vez establecida la carcinogénesis en una clona celular, algunas células de su progenie adquieren mutaciones adicionales o cambios epigenéticos, que les otorgan ventajas para sobrevivir, condición denominada progresión tumoral. La tasa de crecimiento de la población celular dentro de un tumor, depende de cuatro parámetros: tasa de mutación, número de células en proliferación, tasa de proliferación/ apoptosis y tasa de adaptación a condiciones microambientales no adecuadas, para su supervivencia.8
Las células cancerosas adquieren una variedad de propiedades especiales en la carcinogénesis y en la progresión tumoral, una de las más importantes son las alteraciones en las vías de señalamientos intracelulares, capaces de ignorar las señales de su microambiente que normalmente mantienen a la proliferación celular bajo estricto control.8 Estos cambios incrementan la capacidad de que las células tumorales sobrevivan, crezcan y se dividan en el tejido original, y luego metastaticen (sobrevivan y proliferen en tejido diferente). Las células cancerosas no solamente ignoran las señales inhibitorias de crecimiento, sino que ellas continúan su crecimiento, en ausencia de las señales estimuladoras de crecimiento que normalmente requieren. La evolución del tumor no sólo depende de las células tumorales por sí mismas, sino también de las llamadas colectivamente células estromales, ubicadas en el microambiente tumoral.9
La mayoría de las células cancerosas son genéticamente inestables, acumulan cambios genéticos que puede afectar aun más, los sistemas de reparación o replicación del DNA, o los sistemas de mantenimiento en el número e integridad de los cromosomas, previamente dañados. Esta inestabilidad genética aumenta cuando coinciden o se producen cambios simultáneos, que alteran los controles epigenéticos. Como resultado de ello, las células cancerosas experimentan tasas diez a 20 veces mayores de alteraciones en los nucleótidos, que las células normales.2 El crecimiento tumoral depende de defectos en el control de proliferación, muerte y diferenciación celular. En general, las células cancerosas evaden la senescencia replicativa por dos mecanismos: adquieren cambios genéticos/epigenéticos que eluden el control, para detener el ciclo celular en condiciones de telómero corto (por mutaciones que inactivan la vía de p53), o mantienen una alta actividad de telomerasa).10
¿ INICIACIÓN
El desarrollo del cáncer en un individuo requiere muchos pasos, cada uno provocados por múltiples factores (algunos dependientes de la constitución genética del individuo y otros dependientes del ambiente y de su estilo de vida), esta transformación es el resultado de cambios básicos en los señalamientos intracelulares que gobiernan la proliferación y la sobrevivencia celulares. Se ha considerado que el 80% de los cánceres podrían ser evitables o por lo menos, ser pospuestos en su presentación, a través de evitar el tabaquismo, la exposición a radiaciones ultravioletas o a la inhalación de fibras de asbesto, lo cual disminuiría el desarrollo los cánceres broncogénico, de piel y mesotelioma. Diferentes estudios epidemiológicos, han demostrado que un mayor consumo de grasas animales y alcohol en la dieta, aumentan el riesgo de desarrollar cáncer, mientras que el consumo de frutas y vegetales reduce el riesgo.9 Como mencionamos, los carcinógenos incluyen varias sustancias químicas y varios tipos de radiaciones (las UV de la radiación solar y las ionizantes, como los rayos X, y partículas). Ejemplos de carcinógenos químicos, incluyen los hidrocarburos aromáticos y sus derivados como aminas aromáticas, nitrosaminas y agentes alquilantes. La evaluación del poder mutágeno de una sustancia, se puede valorar in vitro con la prueba de Ames. Pocos carcinógenos actúan directamente sobre el DNA, la mayoría lo hacen después de ser procesados metabólicamente en el hígado, por la citocromo p-450 oxidasa. No todas las sustancias que participan en el desarrollo del cáncer son carcinogénos directos o mutágenos (iniciadores del tumor), sino que funcionan como promotores del tumor, como los esteres del forbol (por ejemplo, el acetato del tetradecanoilforbol, el cual activa a la proteína cinasa C, que a su vez activa la vía del fosfatidilinositol).11 Uno de los efectos de las sustancias promotoras del tumor, es inducir la respuesta inflamatoria crónica local, que causa la secreción de factores de crecimiento y de proteasas, las cuales estimulan directa o indirectamente la división celular. En relación a lo anterior, algunas infecciones crónicas por virus, bacterias y parásitos funcionan como agentes promotores de tumores (lo cual sucede en el 15% de todos los cánceres humanos), particularmente algunos virus de DNA, como los papilomavirus (carcinoma cervicouterino), los virus de la hepatitis B y C (carcinoma hepático), el virus de Epstein-Barr (linfoma de Burkit) y algunos virus de RNA, como el virus 1 de la leucemia de células T, el virus del herpes humano-8 (HHV-8, sarcoma de Kaposi), la bacteria Helicobacter pylori (linfomas y probablemente carcinomas gástricos) y el parásito Schistoma haematobium. Algunos agentes químicos ambientales aceleran la tasa de desarrollo y progresión del cáncer, los cuales funcionan como iniciadores y promotores. No obstante, muchos factores que favorecen el desarrollo del cáncer quedan aún por identificar.9
¿ GENES CRÍTICOS EN EL DESARROLLO DEL CÁNCER
El cáncer es una de las principales causas de muerte de adultos en la población mundial, afectando aproximadamente a uno de cada tres individuos. Sin embargo, su desarrollo es una condición infrecuente, ya que a diferencia de otras enfermedades, en las que se requiere la modificación de una gran cantidad de células, el cáncer resulta de la proliferación/transformación incontrolada de una sola célula. El cáncer depende de la acumulación heredada de cambios genéticos/epigenéticos, que una célula transformada transmite a su progenie. Una de las razones primarias, por la cual muy escasas células se transforman en malignas, es que este proceso requiere de la acumulación de múltiples alteraciones genéticas/ epigenéticas, las cuales ocurren en el curso de muchas divisiones celulares sucesivas, y completarlas toma años. El modelo de la inducción de cáncer a partir alteraciones genéticas (hits) múltiples, ha sido demostrado a través de varias líneas de evidencias. Los tumores malignos más frecuentes (mama, colón, próstata, pulmón), que se originan en tejidos epiteliales (90%), muestran una elevada tasa de división celular. Las células de estos tejidos están constituidas por células madre epiteliales o stem cells (proporcionalmente son una minoría), células progenitoras y células diferenciadas. Las stem cells dada su larga vida y su ilimitado potencial de división celular, tienen la oportunidad de acumular las mutaciones que se requieren, para la transformación maligna.2
¿ PROGRESIÓN
Los cambios genéticos que ocurren durante la progresión tumoral se acompañan de cambios histológicos, inicialmente de cambios identificados como pre-cancerosos. Los genes relacionados al cáncer se han identificado principalmente por mutaciones, las cuales aumentan o disminuyen su función en las vías de señalamientos. Los proto-oncogenes (genes normales o wild-type) al mutar incrementan su función, se convierten en oncogenes. Los genes que han perdido su función en el proceso del desarrollo del cáncer, son llamados genes supresores tumorales (GST). La identificación de algunos oncogenes y GST hace 40 años, abrió la era de la epidemiología molecular del cáncer. Los oncogenes codifican proteínas que promueven la pérdida del control del crecimiento, aceleran la proliferación, conducen a inestabilidad génica, evaden la apoptosis y promueven las metástasis. Los mutaciones de los oncogenes y GST tienen efectos finales similares en aumentar la proliferación y sobrevida celulares, y en promover el desarrollo del tumor.8 Los oncogenes son genéticamente dominantes, ya que una sola copia de un gen provoca que la célula exprese el fenotipo alterado. En contraste, la mutación en una copia del GST no modifica el fenotipo, su efecto es recesivo, a menudo se requiere de la mutación de las dos copias del gen para modificar su fenotipo. La mayoría de los tumores contienen alteraciones tanto en GST como en oncogenes (junto con mutaciones en otros genes adicionales como los que codifican moléculas de adhesión celular, proteasas extracelulares, entre otros). Muchos de los oncogenes conocidos, derivados de los proto-oncogenes, juegan papeles muy importantes en la transmisión de señales del proceso de crecimiento celular viajando desde el microambiente extracelular hasta el DNA, otros oncogenes o las mutaciones en GST causan producción inapropiada de factores de transcripción que pueden conducir a la transformación, ejemplos de ellos son MYC, RB, JUN y FOS (estos dos últimos se asocian a AP1 y se unen a secuencias promotoras y facilitadoras -enhancers-de muchos genes). Otros oncogenes codifican factores de crecimiento o sus receptores (PDGF, EGF, EGFR), proteínas G (KRas), proteína-cinasas citoplasmáticas (RAF, SRC) o productos que afectan la apoptosis (BCL-2), entre otros. 11,12
Diferentes estudios experimentales, que emplean la transfección de oncogenes a células normales, permitieron identificar algunos oncogenes como RAS, ya que las colonias transfectadas mostraron propiedades de las células cancerosas (como falta de inhibición de su crecimiento por contacto). Este fragmento de DNA fue aislado y secuenciado, demostrando la versión mutada del proto-oncogen Ras. La secuencia de Ras humano fue muy parecida al gen SRC de los ratones. La proteína Ras es una GTPasa que participa en la transmisión de las señales de los receptores de superficie celular al interior, su mutación forma un proteína hiperactiva que mantiene la transmisión de la señal aun cuando el GTP sea hidrolizado a GDP,13 el proto-oncogen Ras se ha identificado en gran cantidad de tumores.
Los GST codifican proteínas que generalmente inhiben la proliferación celular, mantienen la diferenciación celular, facilitan la adhesión celular, permiten la reparación del DNA, sostienen la forma y la inhibición de crecimiento por contacto celular. La pérdida de su función bloquea éstas regulaciones y contribuyen al desarrollo de muchos cánceres. Los principales GST codifican proteínas intracelulares, que inhiben la progresión del ciclo celular (por ejemplo p16, p53), receptores o tranductores de señales que inhiben la proliferación (receptor Patched), proteínas que detienen el ciclo celular en caso de daño del DNA (por ejemplo p53), proteínas que promueven la apoptosis, y enzimas que participan en la reparación del DNA. Los GST fueron identificados en modelos de síndromes de canceres hereditarios, el primer GST identificado fue el gen RB, en el tumor retinoblastoma. Los pacientes afectados por retinoblastoma presentan deleción de una banda en el cromosoma, usando como referencia esta deleción, fue posible la clonación y secuenciación en este sitio del gen RB. Más tarde, se descubrió que quienes sufren de esta forma hereditaria de enfermedad, presentaban deleción o pérdida de la función por mutación de una copia del gen RB (primer hit), en cada una de sus células somáticas, pero para desarrollar la enfermedad se requiere la inactivación de la otra copia del gen RB (segundo hit). Las bases genéticas del retinoblastoma fueron aclaradas por Alfred Knudson en 1971. Más adelante, otros GST fueron identificados comparando los genomas de las células tumorales con los de las células no-tumorales, del mismo paciente. La primera copia de los GST puede ser inactivada por mecanismos genéticos (deleción, mutación, pérdida de la heterocigosidad), y la segunda copia es comúnmente eliminada por cambios genéticos menos específicos (alteraciones en la segregación del cromosoma, en la recombinación mitótica, haploinsuficiencia, entre otros), o por cambios epigenéticos que lo inactivan permanente e irreversiblemente.14,15 Su pérdida o inactivación es una frecuente característica de muchos cánceres esporádicos. La predisposición hereditaria al cáncer de colon y mama es asociada a mutaciones heredadas de los GST APC y BRCA-1 respectivamente, un alelo del gen se encuentra dañado, y el individuo se mantiene heterocigoto a la mutación, pero si se produjera otra pérdida o inactivación del otro alelo normal en la célula somática, se denomina pérdida de la heterocigosidad (LOH), lo cual favorecería la tumorigénesis.13
Existen diferentes mecanismos por los cuales los proto-oncogenes son convertidos a oncogenes: 1) una pequeña mutación o deleción del gen, puede producir una proteína hiperactiva, o la modificación del promotor conduce a la sobreproducción de ella, 2) la amplificación del gen conduce a la sobreproducción de la proteína (77 genes reportados), y 3) una reubicación del gen por recolocación cromosomal (el gen cerca de una secuencia reguladora o fusionado con otro gen con transcripción activa). En general, diferentes oncogenes responden a los diferentes carcinógenos, generándose anormalidades específicas.
Aun con el uso de metodologías de exploración molecular de alta tecnología en la identificación de oncogenes, como la hibridación genómica comparativa, los microarreglos de DNA y ensayos de interferencia con microRNA, se estima que un número importante de oncogenes y GST candidatos, quedan aún por ser identificados.
Si el objetivo es entender cómo las células tumorales funcionan en el contexto molecular, es necesario saber cómo las proteínas codificadas por los oncogenes y GST, participan en las diferentes vías bioquímicas de regulación.11,13 Esto se ha determinado, gracias a muchos estudios realizados en el desarrollo embrionario y al uso de modelos de ratones modificados con ingeniería genética. La mayoría de los mecanismos de señalamientos moleculares, que funcionan en un cuerpo adulto para mantener la homeostasis, operan el desarrollo embrionario, los cuales dependen de la comunicación célula-célula, y de la regulación del crecimiento, proliferación, diferenciación, muerte, movimiento y adhesión celular. Para estudiar, cómo las diferentes mutaciones de los genes críticos del cáncer afectan los tejidos en un organismo, diversos experimentos en los ratones transgénicos, knockout y knockdown han sido utilizados. Típicamente un ratón transgénico que expresa los oncogenes MYC o Ras, muestran aumento de proliferación celular, y sus células pueden desarrollar tumores. La probabilidad de tumorigénesis se eleva, cuando en modelos genéticamente modificados se expresan dos o más oncogenes. Algo similar sucede cuando dos o más GST son eliminados en ratones knockouts, o cuando se combina la expresión de oncogenes con la supresión de GST.
Las mutaciones o alteraciones epigenéticas de los oncogenes y los GST, corresponden a los "genes conductores" de la carcinogénesis y de la progresión tumoral,16,17 sin embargo en la transformación celular, muchos otros genes sufren alteraciones genéticas que no afectan directamente la carcinogénesis, los cuales han sido llamados "genes pasajeros", que han son modificados secundariamente al proceso de la transformación.
Muchos de los mapas de las principales vías de señalamientos intracelulares, fueron identificados al estudiar los mecanismos de la formación tumoral.13,18-20 Una gran cantidad de proteínas codificadas por proto-oncogenes y GST, incluyen ejemplos de cada uno de los tipos de proteínas involucrados en los señalamientos celulares. Las mutaciones en estos genes alteran los componentes moleculares de modo que crean señales proliferativas aun cuando las células no las requieran, conduciendo al crecimiento celular, replicación del DNA y división celular inapropiada. No es sorprendente que las mutaciones de los genes, que afectan directamente el control central del ciclo celular, se encuentren en muchos cánceres. Las proteínas codificadas tanto por oncogenes y GST, actúan a menudo dentro de una o varias vías de señalamientos. Las vías de señalamientos de los principales procesos celulares oncogénicos, fueron propuestas en un mapa integral por Hanahan y Weinbeg en el 2000.8
En la última década se ha demostrado que diferentes microRNAs participan en la regulación fisiológica, y eventualmente en la carcinogénesis y progresión tumoral, su principal forma de regular es por inhibición de la expresión del mRNA de los GST (actúan como oncogenes), como en la leucemia linfocítica crónica y en algunos linfomas. 21
Cada una de las diferentes biomarcas fenotípicas de los tumores, implica una o varias vías moleculares oncogénicas (modificación de su correspondiente vía molecular fisiológica), de acuerdo a las etapas de carcinogénesis y progresión del tumor.10,13 Muchas de las vías se encuentran entrelazadas, y de igual forma varios oncogenes y GST participan en ellas, así por ejemplo la vía de la proliferación celular se enlaza con la vía del crecimiento celular (que involucra el metabolismo anabólico), y ambas comparten los señalamientos de la vía PI3kinasa/ Akt. De manera similar, la vía de proliferación celular se encuentra estrechamente relacionada con la vía de la apoptosis, y con las que gobiernan la respuesta al daño del DNA, de esta manera cuando una célula tumoral presenta mutaciones del gen p53, es capaz de sobrevivir y proliferar aun cuando el DNA se encuentre dañado.
¿ CARCINOGÉNESIS EN EL CÁNCER COLORRECTAL
Uno de los principales modelos para entender y estudiar la carcinogénesis, ha sido el cáncer colorrectal (CRC), basados en estudios en humanos y en animales experimentales. 20 Los estudios en CRC demostraron las evidencias de la participación de múltiples alteraciones genéticas (hits) en su oncogénesis, debidas a la exposición de carcinógenos colónicos (nitratos, tabaco, alcohol, entre otros). La progresión macroscópica del CRC fue explorada in vivo, mediante el uso de la colonoscopia, lo cual permite obtener material tumoral para el estudio molecular. El modelo de carcinogénesis fue basado tanto en estudios de CRC esporádico, como en CRC hereditario, lográndose asociar los datos micro/macroscópicos con los cambios en genes específicos. El CRC se inicia en el epitelio del colon y recto, donde normalmente estas regiones son renovadas, la capa del epitelio es remplazada completamente en una semana a partir de las stem cells localizadas en las criptas intestinales (señalamientos de organización y control de la proliferación celular). Las mutaciones de la vía Wnt y de los genes que regulan la beta-cateninanuclear, son los mecanismos más importantes que alteran el control de la proliferación celular de las stem cell intestinales (ISC), si estas son sobreactivadas, se puede iniciar la carcinogénesis. Algunos modelos proponen que el incremento de de las ISC, se debe al aumento de expresión de los genes blancos de la vía Wnt (c-MYC, c-JUN, ciclina D) junto con los de la vía Notch, que regulan el switch para que las ISC produzcan mayor cantidad de células progenitoras.22
El CRC es un tumor frecuente en las poblaciones occidentales, 75% de los casos se presentan en forma esporádica (adultos sin predisposición hereditaria), y en 25% con antecedente de familiar de poliposis familiar o CRC hereditario sin poliposis (HNPCC), o eventualmente en pacientes con colitis de Crohn. La exploración del CRC como tamizaje mediante colonoscopia, identifica lesiones pre cancerosas, pequeños tumores benignos, llamados adenoma o pólipos (los cuales se incrementan después de la exposición al carcinógeno), particularmente un tipo de ellos, los pólipos adenomatosos (o adenomas) son precursores del CRC, y la progresión a la transformación es lenta y tarda cerca de diez años. Si durante ese periodo, los pólipos son resecados a través de la colonoscopia, la incidencia de desarrollar CRC baja a menos de una cuarta parte. El estudio histopatológico de los pólipos menores de un cm de diámetro, muestran un epitelio casi normal (mínimas áreas de displasia en diferentes grados). Los pólipos mayores muestran células indiferenciadas, algunas áreas con células relativamente normales alternadas con otras áreas con células cancerosas y/o alteraciones tisulares (mutaciones en K-RAS, inactivación de APC, p53 y DCC). En etapas más tardías de la enfermedad, las células cancerosas se vuelven invasivas, rompen la membrana basal, se diseminan a través de las capas musculares, y finalmente metastatizan por vía linfática a los ganglios linfáticos y por vía sanguínea a otros órganos distantes.
La carcinogénesis del CRC puede desarrollarse a través de cuatro mecanismos:20 la secuencia adenoma-carcinoma, la forma "de novo", la del tipo HNPCC, y la del tipo asociada a la colitis. Una serie de lesiones genéticas, son comunes en todos los mecanismos de carcinogénesis en el CRC, y de ellas destacan las mutaciones del proto-oncogen K-Ras (frecuentemente en el exón 12), y la de los GST p53, APC y DCC (gen frecuentemente deletado en CRC), los dos últimos frecuentemente por LOH. El p53 detiene el ciclo celular induciendo a p21 (proteína que detiene el ciclo celular inducido por daño del DNA-GADD45). Otros más han sido detectados en menor número de CRC, entre los ellos se encuentran las mutaciones de beta-catenina, TGFR-II, Smad4 y MLH1, junto con otros genes de reparación del DNA. El 40% de los CRC esporádicos cursan con mutaciones puntuales de K-Ras, y cerca de 60% cursan con mutaciones o deleciones de p53.23
El CRC hereditario familiar es poco frecuente y se presenta en familias con el Síndrome de Poliposis Adenomatosa Familiar Coli, en quienes se desarrollan cientos a miles de pólipos, en los cuales se ha identificado la deleción o inactivación de una copia del gen APC. Mientras que en el 80% de los CRC esporádico, la otra copia es inactivada por mutaciones adquiridas en el transcurso de la vida (condición similar a lo que ocurre con RB en los pacientes con retinoblastoma). La proteína APC participa en el componente inhibitorio de la vía de señalamiento Wnt, mediante su unión y degradación de beta-catenina, lo cual impide su migración al núcleo, donde normalmente actúa como un regulador transcripcional, para mantener en estado activo a las stem cell de las criptas intestinales. La proteína APC no solo interactúa con la beta-catenina, sino que funciona en la construcción del huso mitótico por microtúbulos, la pérdida de su función favorece diferentes anormalidades cromosómicas.
Algunas familias con CRC hereditarios con poliposis, como las familias con HNPCC, cursan con mínimas anormalidades cromosómicas, pero debido a que presentan mutaciones en los diferentes genes del sistema de reparación de daño del DNA como hMSH2, hMLH1, hPMS1, hPMS2 y hMSH6, les provocan inestabilidad genética en secuencias repetitivas cortas de DNA o microsatélites (cambios en la longitud y frecuencia de repeticiones de nucleótidos/dinucleótidos: AAAA... CACACA...).20
Fearon y Vogelstein en 1990 propusieron un modelo molecular de la carcinogénesis y la progresión tumoral del CRC, basado en la acumulación secuencial de eventos genéticos que se asocian a la transición de adenomas a carcinomas: Epitelio normalàHiperplasia del epitelio (pérdida de APC)àAdenoma temprano (mutación de K-Ras) àAdenoma intermedio (pérdida de DCC)àAdenoma tardío (pérdida de Smad4 y otros GST)à Carcinoma (pérdida de p53)à Invasión y metástasis (activación de otros oncogenes y desactivación de otros GST).
Así los pasos de la carcinogénesis del CRC, sugieren una mutación inicial en los genes de reparación del DNA, y le siguen las alteraciones en los genes que regulan la proliferación celular (lesiones génicas y epigenéticas). Las alteraciones fenotípicas tumorales, logradas en este orden por las mutaciones de estos genes específicos, pueden ser logradas por otras diferentes combinaciones de otros oncogenes y GST.20,22 Globalmente, cada tipo de cáncer cursa con patrones parecidos de alteraciones génicas y epigenéticas acumulables y progresivas, pero cada cáncer en particular a pesar de pertenecer al mismo tipo, presenta un patrón individualizado de lesiones genéticas y epigenéticas.10,23
¿ CONCLUSIONES
La iniciación y progresión del cáncer es un complejo sistema biológico, donde participan alteraciones genéticas, epigenéticas y proteómicas, que conducen al desarrollo de un fenotipo celular tumoral. Cada tipo de cáncer cursa con alteraciones globalmente parecidas, cada individuo con un mismo tipo de cáncer, cursa con alteraciones particularmente específicas.
Correspondencia: Dr. Víctor M. Valdespino.
Calle Andrés Molina Enríquez 361, Colonia Ampliación Sinatel, CP 09479, Delegación Iztapalapa, Ciudad de México.
Teléfono: (55) 5674 3439.
Correo electrónico: vvaldespinog@yahoo.com.mx