


Las células adquieren diversos patrones de expresión génica durante la diferenciación para adaptarse a un entorno cambiante. Las alteraciones epigenéticas y genéticas son consideradas como 2 mecanismos independientes que participan en la aparición y progresión del cáncer. Los mecanismos epigenéticos pueden ser tan importantes para los eventos biológicos como los mecanismos genéticos, que no implican un cambio en la secuencia de ADN, pero si tienen un importante papel en la modificación de la expresión génica. Durante la última década, la investigación en las alteraciones epigenéticas en la progresión del cáncer ha sido mejorada gracias a la aparición de nuevas tecnologías, para buscar aplicaciones en el diagnóstico y la terapia de esta enfermedad. En esta revisión se discute la evidencia actual sobre el papel de los mecanismos epigenéticos en la progresión de varios tipos de cáncer, destacando las ventajas de la investigación de la epigenética en futuros nuevos tratamientos.
The cells acquire different patterns of gene expression during differentiation to adapt to a changing environment. Epigenetic and genetic alterations are considered to be 2 independent mechanisms involved in the onset and progression of cancer. Epigenetic mechanisms may be important for biological events such as the genetic mechanisms that do not involve a change in DNA sequence, but if they have an important role in modifying gene expression. During the last decade, research in epigenetic alterations in cancer progression has been improved thanks to the emergence of new technologies, to find applications in the diagnosis and therapy of this disease. In this review is discussed the current evidence on the role of epigenetic mechanisms in the progression of various cancers highlighting the advantages of the investigation of epigenetics in future new treatments.
Introducción
En los primeros años de la revolución de la Biología Molecular, la investigación del cáncer se ha centrado principalmente en los cambios genéticos (es decir, aquellos que alteran las secuencias de ADN). Aunque esto ha sido de gran utilidad en nuestra comprensión de la patogénesis y la biología del cáncer, hay otro ámbito en el proceso de carcinogénesis que no implica cambio de la secuencia de ADN, denominado "epigenética"1. En 1942, Conrad H Waddington2 acuñó este término. Los procesos epigenéticos son naturales y esenciales para muchas funciones del organismo, pero si ocurren de forma incorrecta, hay efectos adversos en la salud. El notable avance en este nuevo campo ha llamado la atención debido a las posibles aplicaciones de estos nuevos avances en la medicina y diversos campos de la investigación biomédica. El resultado es una apreciación más amplia de los fenómenos epigenéticos en la etiología de enfermedades humanas comunes, en particular el cáncer.
El cáncer por ser un proceso multifactorial complejo que incluye cambios epigenéticos, se ha visto recientemente que estas alteraciones podrían utilizarse como "biomarcadores" para el diagnóstico molecular en varios tipos de cáncer ya que muestran ser seguros en el diagnóstico y el pronóstico, además estas alteraciones podrían ser indicadoras de exposición temprana a determinados cancerígenos potenciales3,4.
En general, las modificaciones epigenéticas son reversibles reordenamientos de la cromatina en las células normales que modulan la expresión de genes, sin cambiar la secuencia de ADN. Las alteraciones de este equilibrio, que afectan principalmente a los mecanismos de la metilación del ADN, hipermetilación, acetilación de histonas, son frecuentemente implicadas en la génesis del cáncer. En las células neoplásicas, la abundancia de las histonas desacetiladas se asocia generalmente con la hipermetilación del ADN y silenciamiento de genes. Varios compuestos antineoplásicos están siendo dirigidos a la histona desacetilasa in vitro5 y a otros mecanismos epigenéticos6-9.
El objetivo de esta revisión es discutir la evidencia actual sobre el papel de los mecanismos epigenéticos en la progresión de varios tipos de cáncer, destacando las ventajas de la investigación de la epigenética en futuros nuevos tratamientos.
Diferencias entre genética y epigenética
Muchos procesos celulares, incluyendo la expresión de genes y la replicación del ADN, a menudo se rigen por mecanismos que entran en la categoría de "la genética clásica". Esto significa generalmente que son controlados por elementos tales como promotores, potenciadores, o sitios de unión para proteínas represoras, que están presentes o ausentes en la secuencia de ADN. Un ejemplo de este tipo de regulación es el control de la expresión de un oncogén. En las células normales (no cancerosas), este gen no se expresa. Sin embargo, en una célula cancerosa, este gen podría haber adquirido una mutación, que es un cambio en la secuencia de ADN, que permite que el oncogén se exprese, y por lo tanto, pueden contribuir a la progresión del cáncer. Además de estos mecanismos de regulación, casi todos los procesos celulares también pueden ser regulados por mecanismos epigenéticos. La palabra "epigenética" literalmente significa "además de cambios en la secuencia genética". Los mecanismos epigenéticos pueden ser tan importantes para los eventos biológicos como los mecanismos genéticos, y también puede dar lugar a cambios estables y heredables. Sin embargo, la gran diferencia entre la regulación genética y epigenética es que los mecanismos epigenéticos no implican un cambio en la secuencia de ADN, pero sí tienen un importante papel en la modificación de la expresión génica (fig. 1), mientras que los mecanismos genéticos implican la secuencia y los cambios o mutaciones del ADN primordial de esta secuencia6.
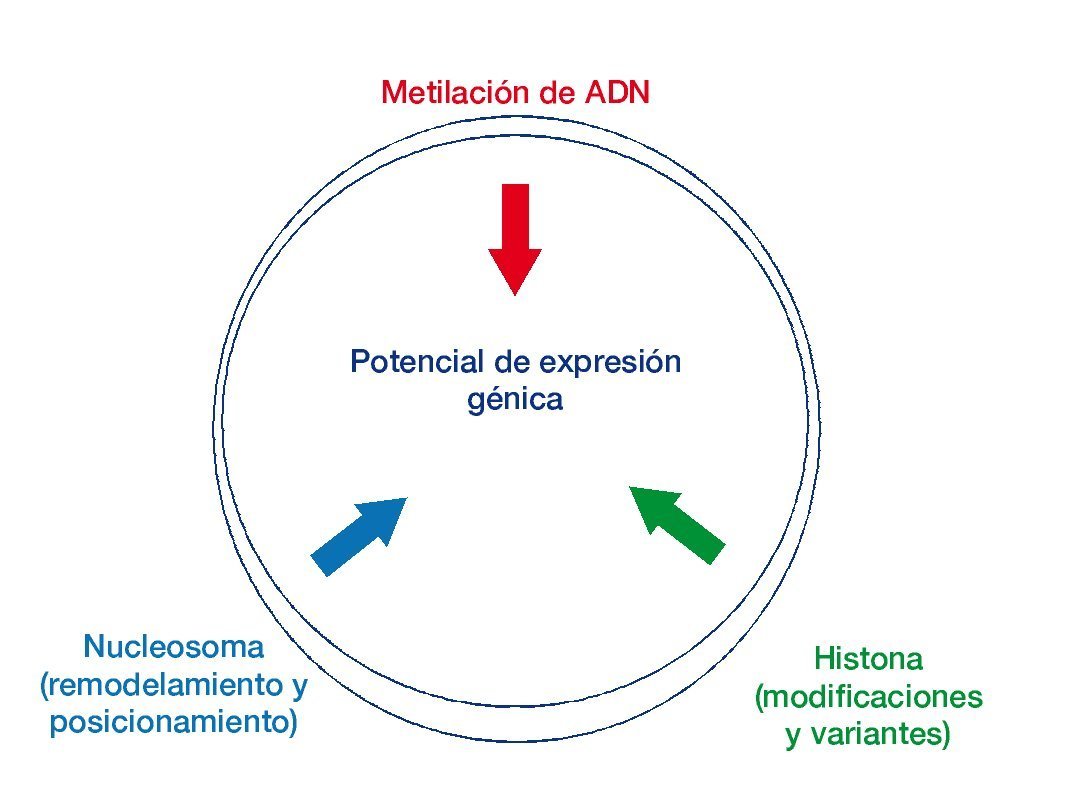
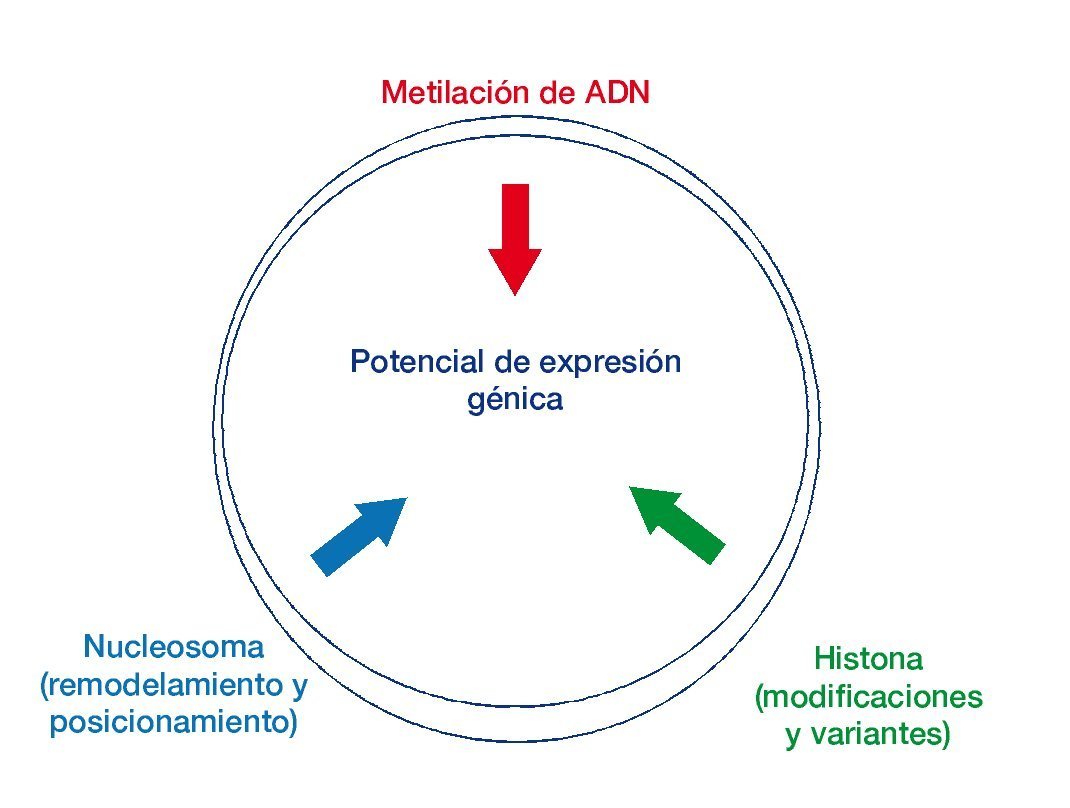
Figura 1 Las mutaciones genéticas con modificadores epigenéticos en el cáncer.
El potencial de expresión génica se ve influenciado por los mecanismos epigenéticos. Se muestra la interacción entre los procesos epigenéticos en la especificación de los patrones de expresión génica7.
Las alteraciones epigenéticas y genéticas son consideradas como 2 mecanismos independientes que participan en la carcinogénesis. La secuenciación completa del exoma de varios tipos de cáncer ha dado como resultado el inesperado descubrimiento de muchas mutaciones, que inactivan a los genes que controlan el epigenoma. Estas mutaciones tienen la capacidad de alterar los patrones de metilación del ADN, modificar las histonas, reubicar a los nucleosomas. La alteración genética del epigenoma contribuye al cáncer al igual que el proceso epigenético puede causar mutaciones puntuales y desactivar los mecanismos de reparación del ADN. Esta interferencia entre el genoma y el epigenoma abre nuevas posibilidades para la terapia contra el cáncer7.
Tumorigénesis y epigenética
La tumorigénesis es un proceso progresivo complejo y multi-factorial de la transformación de células normales en células malignas, se caracteriza por la acumulación de múltiples fenotipos hereditarios específicos de cáncer provocados por eventos mutacionales y/o no mutaciones (epigenética). La acumulación de pruebas sugiere que la exposición ambiental a sustancias naturales, agentes químicos y físicos, tienen un papel crucial en el cáncer humano. En un sentido amplio, la carcinogénesis puede ser inducida a través de mecanismos ya sea genotóxicos o no genotóxicos, sin embargo ambos causan importantes cambios epigenéticos. un ejemplo de alteraciones epigenéticas inducidas por varios agentes carcinógenos químicos, son el arsénico, el 1,3-butadieno, agentes farmacéuticos y biológico8.
Las modificaciones epigenéticas, por ciertos mecanismos, se cree que desaparecen con cada nueva generación, durante la gametogénesis y durante el proceso de desarrollo. Sin embargo, uno de los informes más alarmantes publicadas en 2005 desafía esta creencia y sugiere que los cambios epigenéticos pueden mantenerse al menos 4 generaciones posteriores8-10.
Modificación de las histonas
Las modificaciones de las histonas son uno de los principales mecanismos epigenéticos que regulan la expresión de genes y cuando están desequilibrados conducen a cáncer9. Las histonas ya no son consideradas como proteínas simples de "embalaje de ADN", ya que ellas están sujetas a un gran número de modificaciones postraduccionales incluyendo acetilación, metilación, fosforilación, ubiquitinación, sumoilación (modificación postraduccional implicada en diversos procesos celulares, tales como el transporte nuclear citosólico, la regulación transcripcional, la apoptosis, la estabilidad de proteínas, respuesta a estrés, y la progresión a través del ciclo celular), ribosilación de ADP, deiminación (citrulinación) e isomerización de la prolina. Entre estas modificaciones, la acetilación de histonas y la metilación están relativamente bien estudiadas. La acumulación de pruebas indica que el estado de metilación y acetilación de residuos de lisina o arginina específicos, desempeñan un papel crucial en la regulación de la expresión génica10.
Metilación del ADN
Los cambios epigenéticos son alteraciones en la expresión génica, independientemente de los cambios en la secuencia de ADN. Muchas modificaciones epigenéticas, tales como la metilación del ADN tienen notorios efectos en la expresión génica. La metilación del ADN en las islas cpG silencia la expresión génica al interferir con maquinaria transcripcional9. La metilación del ADN puede interferir en la progresión del ciclo celular y la diferenciación celular, ya que los reguladores del ciclo celular, tales como p16, p21, p27, y p53, son silenciados por la metilación en muchos cánceres9. un ejemplo de ello, es el gen receptor del ácido retinoico (RAR-β2), importante en la diferenciación9.
La metilación del ADN es una modificación epigenética que ocurre en los residuos de citosina en la secuencia 5'-CG-3'. Está bien establecido que la metilación del ADN actúa como un represor transcripcional de la expresión génica, mediante el reclutamiento de proteínas represivas. Éstas incluyen la proteína 1 de unión a Metil-CpG (MeCP1) y proteínas con un dominio de unión a metil, tales como MBD1, MBD2, MBD3, MBD4 y MeCP2. Estas proteínas dificultan la transcripción a través del reclutamiento de otros factores tales como el complejo de remodelación del nucleosoma. En el caso de MecP2 con capacidad de unirse a una sola citosina metilado simétricamente y contribuyendo a represión de la transcripción a largo plazo. La unión de estos factores proteínicos conduce a la condensación de ADN y le confieren estabilidad al cromosoma.
Este proceso de metilación del ADN es mediado por las enzimas denominadas ADN metiltransferasas (DNMT). La DNMT3a y DNMT3b son responsables de la metilación durante la embriogénesis. DNMT1 se ha caracterizado como la metiltransferasa que mantiene la metilación del ADN entre divisiones celulares, siendo altamente expresada en células de cáncer9.
Recientes estudios de secuenciación de genomas de algunos tipos de cánceres humanos, han identificado mutaciones específicas del tumor en los genes que codifican proteínas que funcionan en la regulación de la cromatina, aunque su importancia funcional no siempre ha sido clara. un estudio realizado por Zhu et al.11 ha identificado mutaciones en la histona-lisina-n-metiltransferasa SETD2, demostrando que éstas cooperan con otras aberraciones genéticas en la leucemia12.
La metilación del ADN puede contribuir al desarrollo de la resistencia endocrino adquirida, ya que la ablación hormonal es el tratamiento de elección para los tumores de mama sensibles a las hormonas, pero hasta el 40% de los pacientes inevitablemente recaen, y estos tumores refractarios de la hormona a menudo tienen un mal pronóstico. Como alternativa, los tratamientos deben centrarse en seleccionar subpoblaciones resistentes con estas alteraciones epigenéticas13.
La metilación reversible de citosinas puede ser medida con precisión por diversos métodos moleculares y patrones de metilación del ADN, ya que están vinculados a importantes vías tumorigénicas. Los cambios en la metilación clínicamente relevantes son conocidos en cánceres humanos comunes tales como de cuello uterino, próstata, mama, colon, vejiga, estómago y pulmón. La metilación diferencial puede tener un papel central en el desarrollo y el resultado de la mayoría de los tumores malignos humanos. El advenimiento de la secuenciación profunda representa una gran promesa para epigenómica, con herramientas bioinformáticas listas para revelar un gran número de nuevos objetivos para el pronóstico y la intervención terapéutica14.
Hipermetilación del ADN
En el cáncer hay un número de genes con hipermetilación aberrante que se asocian con la recurrencia de la enfermedad. Es evidente que la hipermetilación aberrante inactiva los genes relacionados con el control del ciclo celular, apoptosis y la reparación del ADN (por ejemplo, la pérdida de expresión MLH1 tiene un papel importante en la carcinogénesis, estos hallazgos sirven como enfoques para la prevención, el diagnóstico, la evaluación de riesgos y el tratamiento de la enfermedad). Los análisis para la detección tienen alta sensibilidad para la identificación de las células cancerosas en muestras de esputo, sangre y biopsia. Hay muchos intentos de utilizar inhibidores de la metilación como agentes contra esta patología, y las anormalidades epigenéticas útiles como biomarcadores de la sensibilidad a los fármacos contra el cáncer, para identificar las características biológicas de las células tumorales y así determinar las mejores opciones de tratamiento basadas en la hipermetilación. Por ejemplo, la hipermetilación aberrante del gen CHFR se correlaciona con la sensibilidad celular a los inhibidores de microtúbulos, esto puede ser útil en el tratamiento de cáncer endometrial tipo I. Un objetivo primordial de la epigenética es identificar el tipo de metilación hereditaria responsable del cáncer para mejorar el diagnóstico y el tratamiento basado en el control de la metilación15.
Epigenética en algunos tipos de cáncer
Cáncer de pulmón
Varios estudios han demostrado que la metilación de los genes supresores de tumores conduce a la expresión inactivación de genes y presenta un importante mecanismo de desarrollo de tumores. PR (PRDI-BF1 y RIZ) las proteínas de dominio PR (PRD1-BF1 y RIZ) denominadas PRDM, son una familia de factores de transcripción de tipo Kruppel, de los cuales 17 son conocidos actualmente en el cuerpo humano. La evidencia actual sugiere que los miembros de la familia PRDM juegan un papel importante en la diferenciación celular y transformación maligna, ya que actúan como supresores de tumores. Para esto, la sustancia 5-aza-2-dC reduce la metilación de genes de PRDM2, PRDM5 y PRDM16 en las líneas celulares de adenocarcinoma de pulmón A549 y HTB-182, disminuyendo subsecuentemente su crecimiento. En la línea de carcinoma de células escamosas de pulmón SK-MES-1, este medicamento también ha sido probado con los mismos resultados16. Consistentemente, 5-aza-2dC aumenta los niveles de mARN y expresión de las proteínas de PRDM2, PRDM5 y PRDM1617.
Cáncer gástrico
En el cáncer gástrico, la infección por Helicobacter pylori (H. pylori) es una causa de la acumulación de la metilación en la mucosa aparentemente normal. Además la infección por el virus de Epstein-Barr es otro inductor de metilación, que conduce a cáncer gástrico18.
Cáncer colorrectal
El cáncer colorrectal (CCR) es muy heterogéneo, éste implica varias vías moleculares. Además, también se ha evaluado que la detección de metilación de ADN para el diagnóstico y pronóstico19. La metilación aberrante del ADN es una alteración epigenómica común en esta carcinogénesis. En la carcinogénesis colorrectal, la acumulación de altos niveles de metilación en combinación con la mutación BRAF son característicos18. Las modificaciones de histonas en el CCR son acetilación/desacetilación y metilación/desmetilación de lisina y arginina, residuos dentro de las colas de las histonas. Si bien, la dimetilación y trimetilación de la lisina de la histona H3 (H3K4me2/me3) y acetilación de los aminoácidos de H3/H4 (H3K9ac y H4K9Ac) constituyen marcas transcripcionalmente activas, promotores de genes transcripcionalmente inactivos se caracterizan frecuentemente por trimetilación de la lisina 9 y 27 de la histona H3 (H3K9me3 y H3K27me3). Estas modificaciones de las histonas bivalentes están mediadas por represores transcripcionales, las proteínas del grupo Polycomb (PRC) son instrumentales en el silenciamiento de un grupo específico de genes supresores de tumores en cánceres humanos. Dos complejos represivos PRC multiméricos, PRC1 y PRC2, pueden silenciar los genes, ya sea de forma independiente o sinérgica. El complejo PRC2 es responsable de iniciar la metilación de la histona H3 (H3K27me2/3) a través de sus subunidades enzimáticas EZH1 y EZH2, mientras que el complejo PRC1 está involucrado en el mantenimiento del silenciamiento de H3K27me2/3, así como la posterior monoubiquitinación de la Lys 119 en la histona H2A (H2AK119ub) a través de la RING1A ubiquitina ligasa RING1A y RING1B. Mientras que el núcleo del complejo multimérico PRC2 se compone de 4 componentes EZH1/2, SUZ12, EED y RbAp46/48 (o RBBP7/4), pero la composición de complejos PRC1 es más variable con sólo 2 componentes comunes básicos: RING1A/B, junto con IMC1, HPC/CBX, HPH y YY1. El silenciamiento transcripcional mediado por PRC, se ha planteado la hipótesis de que juega un papel en el CCR, debido a que muchos genes que están frecuentemente hipermetilados en el CCR son objetivos del grupo PRC. Ambas proteínas PRC1 y PRC2 interactúan con las metiltransferasas de ADN (DNMT1 y DNMT3b), se establece un papel clave potencial de estas proteínas en la catálisis de la metilación asociada al silenciamiento transcripcional de genes diana en las células cancerosas. También, EZH2 se sobreexpresa frecuentemente en los CCR y predice una mejor supervivencia libre de recurrencia en pacientes con CCR. Además, el agotamiento intracelular de EZH2 induce la detención del ciclo celular, inhibe el crecimiento celular de células de CCR y conduce a la reducción de la expresión de varios genes asociados con el cáncer implicados en la proliferación o invasión incluyendo Dag1 (codifica a distroglicano), MageD1 (familia de antígenos del melanoma D1), SDC1, TIMP2 y Tob11,18.
En el CCR la deficiencia de folato (vitamina B9) puede causar carcinogénesis por medio de la inducción a hipermetilación de genes específicos y la hipometilación global de genoma20. La pérdida de imprinting (LOI) implica pérdida del patrón normal de expresión de un alelo parental específico, y en el cáncer conduce a la activación de genes impresos de promoción del crecimiento tales como el factor de crecimiento similar a la insulina II (IGF2), así como también el silenciamiento de genes supresores de tumores tales como p57KIP2 y ARH1. La LOI se puede producir en la mucosa colónica de los pacientes con CCR en sus tumores. Las alteraciones epigenéticas son simplemente consecuencias finales de la neoplasia. Esta LOI es vinculada a los casos que muestran inestabilidad de microsatélites (MSI) en los tumores21.
Este tipo de cáncer ha sido usado ampliamente como modelo para investigar la expresión génica aberrante. La radiación ionizante (IR) y otros agentes cancerígenos inducen a cambios en la expresión génica, interrupción de la detención del ciclo celular y apoptótica in vitro e in vivo22, y de alguna manera servirá para estudiar en más detalle los cambios epigenéticos implicados.
Cáncer próstata
El cáncer de próstata (CaP) es una de las neoplasias humanas más comunes, surge a través de alteraciones genéticas y epigenéticas. Estas alteraciones epigenéticas proporcionan valiosas herramientas para el manejo de pacientes con CaP y se busca usarlos como blanco de los compuestos farmacológicos que reviertan la neoplasia. El potencial de los cambios epigenéticos en el CaP requiere una mayor exploración y validación para permitir la traducción a la clínica23.
Shaikhibrahim et al. (2013)24 ha determinado genes relacionados con procesos epigenéticos tanto en tejidos y líneas celulares metastásicas de CaP. En tumores moderadamente diferenciados los genes TDRD1, IGF2, DICER1, ADARB1, HILS1,GLMN y TRIM27 se sobrerregulan, mientras que TNRC6A y DGCR8 están poco regulados. Pero en tumores pobremente diferenciados, TDRD1, ADARB y RBM3 están sobrerregulados y DGCR8, PIWIL2, BC069781 poco regulados.
En el cáncer, la acetilación de histonas se correlaciona con la activación transcripcional y la desacetilación de histonas está relacionada con el silenciamiento de genes. Por ejemplo en el CaP, el nivel de la histona H3 acetilada (H3Ac) aumenta la expresión tanto del antígeno específico de próstata (APE) potenciador y promotor tras el tratamiento con andrógenos (AR), en paralelo con la acumulación de los niveles de ARNm de APE en la línea celular de CaP LNCaP. El tratamiento a las células LNCaP con un inhibidor de histona desacetilasa tricostatina (TSA), promueve la ARN polimerasa II y la estabilidad de H3Ac en regiones reguladoras de APE para aumentar la transcripción de AR. La acetilación de la histonas está mediado por histona acetiltransferasas (HAT), varios de los cuales han sido caracterizados como co-activadores de AR. Por ejemplo, la CBP y P300 (KAT3A y KAT3B) son reclutados para las regiones reguladoras de APE después del tratamiento de AR y aumentan la transcripción mediada por AR en las células LNCaP9,25.
En contraste con la acetilación de histonas, la metilación de las histonas en la arginina y la lisina se asocia, ya sea con la activación o represión de genes. En el caso de la mono, di y trimetilación de H3K9 (H3K9me1, H3K9me2 y H3K9me3), se relaciona con la represión de los genes diana de AR en las células LNCaP. Por el contrario, la mono y dimetilación de H3K4 (H3K4me1 y H3K4me2) están asociada con la activación de genes mediada por AR en líneas celulares y tejidos CRPC10,25-28.
El carcinoma de próstata se caracteriza por el silenciamiento del gen de la glutatión S-transferasa de clase P1 (GSTP1), que codifica una enzima desintoxicante. El silenciamiento de GSTP1 se produce en la gran mayoría de los casos de alto grado de neoplasia intraepitelial prostática (NIP)25. El silenciamiento epigenético del gen de GSTP1 (> 90%) es una alteración común el CaP26.
Los depsipéptidos tienen un papel muy importante en la reversión de la hipermetilación del ADN y las modificaciones de histonas represivas (reducción de H3K9me2/3 y H3K-27me2/3; aumento de H3K18Ac), induciendo con ello la reexpresión de ARNm de GSTP1, también participa en la inducción a la apoptosis en líneas celulares de CaP (no detiene el ciclo celular).
Se han hecho esfuerzos dirigidos a la metiltransferasa del ADN y las histonas deacetilasas (HDACs) en el CaP y otros tumores sólidos, pero no han tenido el éxito que se vio en las neoplasias hematológicas. Los agentes orales, menos tóxicos, y más específicas se están desarrollando en los tumores sólidos, incluyendo CaP. Las combinaciones de agentes epigenéticos solos o con un agente dirigido, como inhibidores de la señalización del receptor de AR son enfoques prometedores27.
Los mecanismos epigenéticos, especialmente la metilación del ADN diferencial a regiones de control de imprinting (denominadas DMR´s) (fig. 2), normalmente garantizan la expresión exclusiva de los genes improntados de un alelo parental específico. Hay un expresión disminuida de manera significativa de PLAGL1/ZAC1, MEG3, NDN, CDKN1C, IGF2 y H19, sin embargo LIT1 se sobreexpresa significativamente. El gen PPP1R9A, se sobreexpresa fuertemente, bialélicamente en tejidos prostáticos benignos y cancerosos. La expresión de muchos de estos genes se correlaciona fuertemente, lo que sugiere una corregulación, similar a lo que se ha reportado en ratones. Esto indica que un grupo de genes impresos coordinadamente se desregulan en los CaP, independientemente de los cambios de metilación del ADN28.
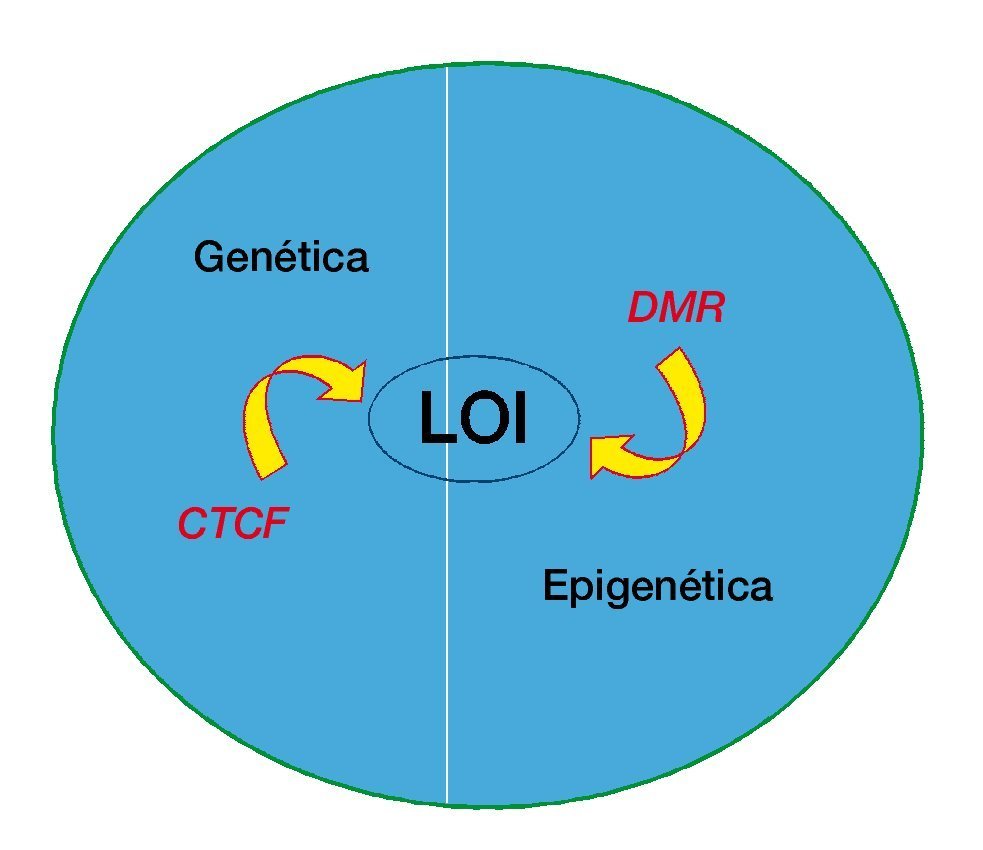
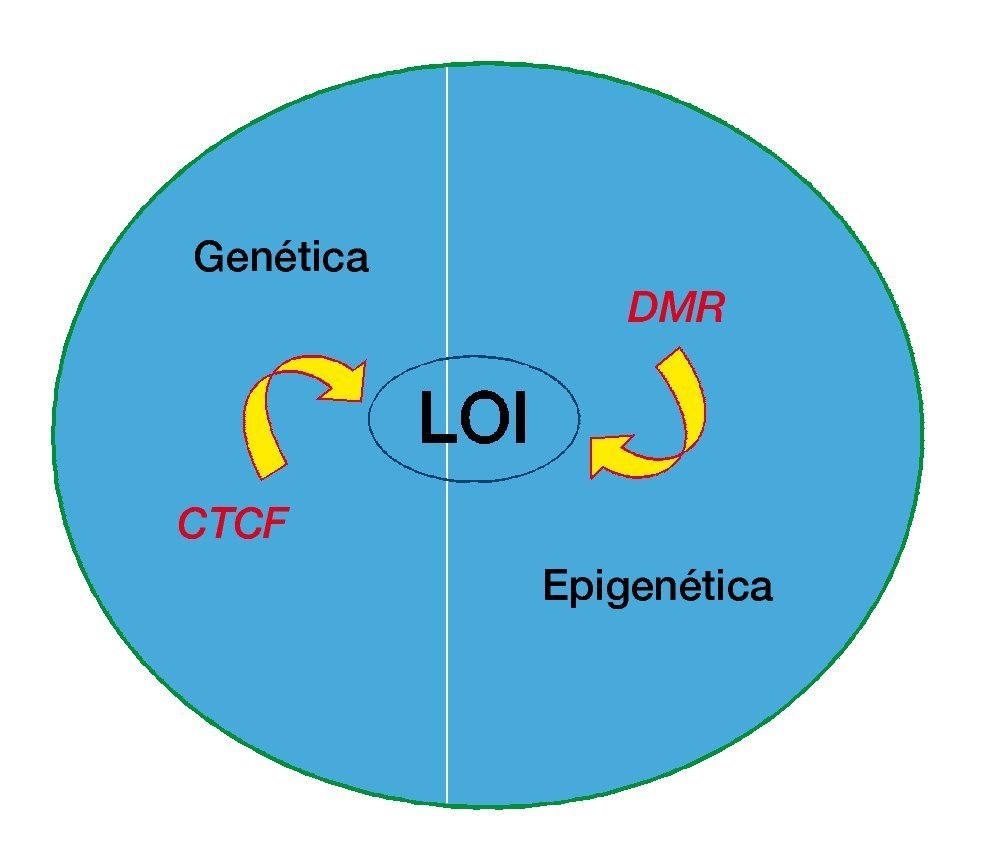
Figura 2 La interrelación de la genética y la epigenética del cáncer. La pérdida de imprinting podría ser causada por la alteración genética de CTCF (factor vinculante a CTCC) o por metilación alterada de las regiones diferencialmente metiladas (DMR).
Desregulación epigenética comprende la hipermetilación y hipometilación del ADN, la sobreexpresión del potenciador de homólogo Zeste 2 (EZH2) y patrones alterados de modificaciones de las histonas. Estos mecanismos aseguran la expresión monoalélica específica de los padres de al menos 62 genes impresos. Aunque es, por tanto, tentador especular que la desregulación epigenética puede extenderse a los genes impresos, los cambios de expresión en CaP sólo están bien documentados para el factor de crecimiento insulínico tipo 2 (IGF2). Los estudios de la literatura y la base de datos sobre los genes impresos en el CaP sugieren que la expresión de mayoría de los genes impresos se mantiene sin cambios, a pesar de las perturbaciones globales en mecanismos epigenéticos. En lugar de ello, los cambios genéticos y epigenéticos selectivos parecen conducir a la inactivación de una subred de genes impresos, lo que podría funcionar en la próstata para limitar el crecimiento celular inducida a través de la vía PI3K/Akt, modular las respuestas de AR y regular la diferenciación29.
Cáncer de mama
Aunque el cáncer de mama es una enfermedad heterogénea, es un reto caracterizar y tratar, la reciente exPLoSión de la investigación genética y epigenética. Los miARN-12b, miARN-145, miARN-21, y miARN-155 significativamente son reducidos en su actividad de expresión génica en múltiples subtipos de cáncer de mama30.
Se sabe que el TGF-β es necesario para la transición epitelial-mesenquimal, y se expresa durante las primeras etapas de la metástasis. Sin embargo, en las células de cáncer de ovario en etapa IV, se observó que el TGF-β fue silenciado por metilación. Debido a que el TGF-β es también conocido por la inhibición del crecimiento, es natural que cuando finaliza el progreso y la diferenciación de las células metastásicas, necesita un rápido crecimiento e invasión, entonces la expresión de TGF-β debe ser disminuida. De esta manera, se requiere el TGF-β durante las etapas iniciales de la metástasis para poder promover la diferenciación, pero debe ser disminuida su expresión durante las etapas posteriores de la metástasis para reducir la inhibición del crecimiento. La doble función de TGF-β es la evidencia de que los diferentes niveles de metilación como la progresión de la metástasis, proporcionan información importante sobre un nuevo blanco terapéutico30.
Los patrones de metilación aberrante del ADN son asociados con muchos tipos de cáncer. La hipermetilación de los genes supresores de tumores (TSG´s) conduce a la inactivación transcripcional, seguida por el silenciamiento de genes y carcinogénesis. En la última década también se ha descubierto que los microARNs (miRNA), ARNs endógenos no codificantes con 19 y 25 nucleótidos juegan un importante rol en varios procesos celulares, incluyendo el crecimiento celular, diferenciación y apoptosis, que contribuyen al desarrollo y progresión de cáncer. Por otra parte, los recientes estudios reportaron que los miARN están involucrados en los cambios en la metilación del ADN. Los cambios genéticos tales como las mutaciones o deleciones se traduce en permanente pérdida de expresión de ciertos genes, mientras que los cambios epigenéticos son a menudo reversibles. La hipermetilación reversible del silenciamiento de TSCs o miRNAs está cada vez más orientada a terapia y prevención del cáncer. Sin embargo, estos retos son particularmente atractivos porque los inhibidores de la inhibición de la metilación del ADN son considerablemente menos tóxicos en los tejidos no cancerosos comparados con otros medicamentos anticancerígenos. El 5-aza-2´-deoxicitidina (DAC), ha sido aprobado por la Administración de Alimentos y Medicamentos (por sus siglas en inglés, FDA, Food and Drug Administration) para el tratamiento de pacientes con síndrome mielodisplásico y leucemia; ya que DAC es uno de los nucleótidos análogos que es activado vía fosforilación por las quinasa de deoxicitidina y es incorporado en el ADN, dando como resultado el agotamiento de la actividad de la metiltransferasa y la desmetilación del ADN31.
Las antraciclinas (doxorrubicina, daunorrubicina, epirrubicina, etc.) son la clase más importante de medicamentos contra el cáncer de mama. Sin embargo, las ventajas terapéuticas se ven afectadas significativamente por la cardiotoxicidad potencialmente mortal y otros efectos secundarios letales. La cardiotoxicidad causada por la doxorrubicina es dependiente de la dosis y acumulativa. La administración repetida de este medicamento origina resistencia por ciertas células dentro de la neoplasia. Una vez que desarrollan resistencia a los medicamentos, estos se vuelven ineficaces a las dosis tolerables habituales. Es por eso que se ha propuesto el efecto altamente sinérgico del tratamiento secuencial con los fármacos anticáncer y epigenéticos (decitabina, DAC; un agente desmetilador y el ácido hidroxámico suberoilanilida [SAHA]; un inhibidor de desacetilación de histonas) para superar la resistencia a fármacos en las células de cáncer de mama; en éste existe mediación por la activación de la expresión del gen p21 que conduce a la detención del ciclo G2/M en un 90%32.
Perspectivas
Alteraciones epigenéticas, tales como modificaciones en los patrones de metilación del ADN y las modificaciones postraduccionales de las colas de las histonas, son fácilmente reversibles por "fármacos" epigenéticos tales como inhibidores de transferasas e inhibidores desacetilasas de histonas. Dado que las alteraciones epigenéticas en las células de cáncer afectan a prácticamente todos los caminos celulares que se han asociado a la tumorigénesis, no es sorprendente que los fármacos epigenéticos muestren actividades pleiotrópicas, capaces de restaurar de forma concomitante la expresión defectuosa de genes implicados en el control del ciclo celular, apoptosis, señalización celular, invasión de células tumorales y metástasis, angiogénesis y reconocimiento inmunológico33.
Las terapias epigenéticas pueden jugar un papel destacado en el futuro tratamiento de los tumores sólidos. Esta posibilidad se basa en la eficacia clínica de los medicamentos existentes en el tratamiento de neoplasias hematopoyéticas definidas, junto con nuevos datos prometedores de los estudios preclínicos y clínicos que examinan estos agentes en los tumores sólidos. Se sugiere que los fármacos actuales representan un enfoque terapéutico específico para la reprogramación de las células de tumores sólidos, una estrategia que debe ser perseguida con la exPLoSión en el conocimiento sobre las bases moleculares del epigenoma de células no cancerosas y cancerosas34.
En los linfomas no Hodgkin (LNH) comprenden un grupo grande y diverso de tumores de origen de los linfocitos, con características moleculares heterogéneas y manifestaciones clínicas. Las terapias actuales se basan en la quimioterapia estándar, inmunoterapia, radioterapia o trasplante de células madre. El descubrimiento de las mutaciones recurrentes en las enzimas que participan en mecanismos epigenéticos, proporciona a los investigadores una base para desarrollar nuevos inhibidores dirigidos a estos enzimas. Varios estudios clínicos y preclínicos han demostrado la eficacia de los fármacos epigenéticos en tratamiento para el LNH y algunos inhibidores específicos que ya han sido aprobados para el uso clínico tales como los inhibidores de DNMT (5-aza-citidina, Vidaza®; 5-aza-deoxicitidine, Dacogen®), inhibidores de HDAC (vorinostat, Zolinza®; romidepsin, Istodax®), inhibidores de HMT, inhibidores BRD (JQ1 dirigida a las proteínas BET de este grupo; está en fase preclínica)35.
Se ha creado un repositorio de información sobre epigenómica en el Centro Nacional de Información Biotecnológica (NCBI) para acceso al público (www.ncbi.nlm.nih.gov/epigenómica), en donde se podrán revisar los últimos avances en este campo de investigación36.
Los patrones de expresión de miARNs se pueden asociar con algunas neoplasias, pero se necesitan estudios adicionales para desarrollar marcadores de diagnóstico y pronóstico muy seguros. Las áreas de la investigación del cáncer (la epigenética, el metabolismo y la vía mTOR, la muerte celular y el sistema inmunológico, la oncología clínica), servirán en un futuro para discutir la aplicación de la epigenética en medicina personalizada del cáncer37.
Terapia epigenética
A diferencia de los cambios genéticos, que son esencialmente fijos, los cambios epigenéticos son intrínsecamente reversibles, y una alteración de la expresión de genes puede activar y desactivar la célula. Esto los hace candidatos atractivos para la intervención terapéutica. Además, existe evidencia creciente que apoya la hipótesis de que las alteraciones epigenéticas pueden ser una fuerza impulsora de resistencia a los medicamentos en el cáncer humano, un fenómeno que se ha reportado en muchos tumores sólidos, incluyendo células de CCR. En consecuencia, 2 clases de compuestos químicos que incluyen los inhibidores de DNMT y HDAC han sido objeto de importantes investigaciones preclínicas y se prueban actualmente para la eficacia en el tratamiento de diversos cánceres humanos en varios ensayos clínicos. Por ejemplo, los medicamentos desmetilantes de ADN 5-azacitidina y 5-aza-2'deoxycitidine (decitabina), ya se utilizan clínicamente para diversos tumores malignos humanos, incluyendo el síndrome mielodisplásico. Estos fármacos actúan a través de su capacidad de incorporarse en el ADN y actuar mediante la prevención de la resolución de una reacción covalente intermedia que atrape e inactive a DNMT, dando como resultado el rápido agotamiento de DNMT y desmetilación concomitante con la replicación del ADN en su curso normal. Estos medicamentos tienen potentes actividades in vitro y algunas respuestas se alcanzan clínicamente, pero la actividad de desmetilación es no específica y las toxicidades son considerables38.
Siete clases de inhibidores de la HDAC han sido desarrollados hasta el momento. La inhibición de estas enzimas lleva a la acetilación de las histonas, que es seguido por una serie de procesos celulares que impactan al crecimiento celular y promueven la tumorigénesis. El medicamento vorinostat también se ha utilizado clínicamente, pero las HDAC son abundantes, y sus papeles terapéuticos en el cáncer aún no se comprenden del todo. Estudios in vitro e in vivo muestran que el vorinostat puede regular a la baja la expresión de la timidilato sintasa en el nivel de transcripción, originando la actividad antitumoral sinérgica cuando se combina con 5-FU en las células de CCR. Dada la estrecha colaboración entre la metilación del ADN y las modificaciones de las histonas para inhibir la transcripción de los genes supresores de tumores, otra estrategia es combinar los inhibidores de HDAC y DNMT, lo que podría tener un efecto más sinérgico en desmetilar genes epigenéticamente silenciados. El tratamiento de combinación con 5-azacitidina y ácido valproico en un ensayo clínico de fase 1 de los pacientes con tumores sólidos refractarios (incluyendo CCR), dio lugar a una disminución significativa en la metilación del ADN total e induce a la desacetilación de histonas con una enfermedad estable que dura hasta 12 meses en un subconjunto de pacientes38-40.
Aunque esto es actualmente un campo emergente, dada la ubicuidad de la hipermetilación en un subconjunto de CCR, la oportunidad de descubrir el uso adecuado de estos fármacos en esta enfermedad es evidente. El desafío será encontrar modificadores epigenéticos eficientes contra tumores sólidos, y otros. Además, hay varios agentes de origen natural "botánicos" tales como curcumina y ácido boswélico que son probablemente seguros (porque se han utilizado durante siglos como especias de alimentos), y estos pueden ser útiles como agentes que previenen el cáncer o como adyuvantes a la quimioterapia convencional. Los ensayos clínicos están estudiando la seguridad y eficacia de diversos fármacos epigenéticos individualmente y en combinación con los fármacos quimioterapéuticos, los cuales revelarán su verdadero potencial clínico. Los autores especulan que las terapias epigenéticas en una variedad de escenarios están al borde de entrar a tallar para la aplicación conjunta con otras terapias actuales.
Conclusión
La complejidad del genoma está regulada por mecanismos epigenéticos heredables, que proporcionan la base para la diferenciación, el desarrollo y la homeostasis celular. La evidencia sugiere que en el cáncer aparece una variedad de cambios epigenéticos, que se producen en las primeras etapas de la enfermedad y mutaciones genéticas paralelas. Los cambios de la metilación del ADN pueden servir como marcadores de diagnóstico, pronóstico y como blancos terapéuticos. La reversibilidad de los cambios epigenéticos hace que sea posible tratar algunos tipos de cáncer con inhibidores de metiltransferasa de ADN e inhibidores de histona desacetilasa. Sin embargo, los agentes demetilizadores disponibles son globalmente eficaces.
Conflicto de intereses
El autor declara no tener ningún conflicto de intereses.
Financiamiento
El autor no recibió patrocinio para llevar a cabo este artículo.
* Autor para correspondencia:
Correo electrónico: luisferscr@gmail.com (Luis Fernando Tume-Farfán).