


¿ INTRODUCCIÓN
A todo tumor maligno originado de células mesenquimatosas se le denomina sarcoma. Las células mesenquimatosas normalmente maduran hasta diferenciarse en músculo esquelético, músculo liso, tejido adiposo, tejido conectivo, hueso y cartílago. El término rabdomiosarcoma (RMS) determina un tumor originado de células mesenquimatosas inmaduras que guardan diferenciación a músculo estriado a pesar de que a menudo se origina en sitios donde no se forma ordinariamente (por ejemplo: vejiga).1,2
El RMS infantil es el tumor maligno de tejido blando más frecuente; representa aproximadamente 3.5% de los casos de cáncer en niños de 0 a 14 años de edad, y 2% entre adolescentes y adultos jóvenes entre 15 a 19 años de edad en Estados Unidos. Su incidencia de es de cuatro a 4.8 casos nuevos por millón, por año en menores de 20 años de edad.3,4 En México se calcula su incidencia anual promedio de 2.5 por millón y la proporción varón:mujer es de 2:1. En el Instituto Nacional de Pediatría (INP) ocupa el séptimo lugar del total de las neoplasias malignas.5 Su distribución geográfica es mundial; no existe una predilección por país y la raza blanca predomina sobre la negra. En lo que se refiere al sitio primario de la enfermedad, éste comprende las siguientes: Vías genitourinarias la relación V:M es de 3.3:1 en neoplasias primarias de vejiga y próstata, y de 2.1:1 en otros sitios de las vías genitourinarias, como vagina. En las extremidades, la relación es de 0.79:1; en la órbita de 0.88:1. En relación con la edad, aproximadamente 87% de los individuos con RMS es menor de 15 años de edad y 13% tiene entre 15 y 21 años de edad. El RMS casi nunca afecta a la población adulta. Al igual que el género, hay diferencias relacionadas con la edad y el sitio primario de la enfermedad: en la vías genitourinarias en pacientes con tumor primario de vejiga y de próstata 75% es menor de cinco años de edad, en la órbita 42% de los sujetos tienen entre cinco y nueve años de edad y en las extremidades 80% es mayor de 10 años de edad. En cuanto al tipo histológico la variedad alveolar es predominante en las extremidades. La variedad botrioides es la única forma de presentación en vagina y vejiga y es casi exclusiva de lactantes.6 Los sitios primarios más comunes donde aparece el RMS son la cabeza y el cuello (por ejemplo el parameníngeo, órbita, faríngeo, etc.), el conducto genitourinario, y las extremidades. Otros sitios primarios menos comunes son el tronco, la pared torácica, abdomen (incluyendo el retroperitoneo y tracto biliar) y la región de perineo/ano.10,11
La mayoría de los casos de rabdomiosarcomas se presentan de forma esporádica, sin ningún factor de riesgo o de predisposición reconocible a pesar de que una pequeña porción de estos, están relacionados con factores genéticos.7
Durante la década de los sesentas, menos de un tercio de niños con RMS sobrevivían después de una terapia local ya fuera resección quirúrgica y/o radioterapia (RT) con o sin monoterapia, con extensas cirugías mutilantes con gran morbimortalidad para el paciente.8 En la época actual, el RMS es curable en la mayoría de los niños que reciben terapia de modalidad combinada, con una supervivencia a cinco años de más de 70% después del diagnóstico. Este logro terapéutico se ha debido a la creación de grupos de estudio interinstitucionales a todo lo largo de países como Estados Unidos, donde uno de los grupos que más avances ha proporcionado en el conocimiento del RMS es el Intergroup Rhabdomyosarcoma Study (IRS) formado en 1972 con el patrocinio del Instituto Nacional de Cáncer (NCI) ante la necesidad de unir resultados que pudieran ser más concluyentes tomando en cuenta que tanto la biología como la clínica des este tumor es muy heterogénea si se toman en cuenta sus múltiples sitios primarios, variedades histológicas y extensión de la enfermedad al diagnóstico, haciendo poco concluyente y de difícil interpretación los resultados de estudios pequeños, desde la creación del IRS se han realizado cuatro estudios consecutivos ( IRS I de 1972 a 1978, IRS II de 1978 a 1984, IRS III de 1984 a 1991, IRS IV 1991 a 1997)9-13 y ya en etapas finales el IRS V iniciado en el año 1998.14
La identificación de factores pronósticos y la adaptación de esquemas de tratamiento de acuerdo a grupos ha sido la mayor aportación de los estudios del IRS que se ha reflejado en el incremento de la sobrevida, disminución de la toxicidad, logrando identificar categorías que requieren mayor intensidad en la terapéutica.
El pronóstico para un niño o adolescente con rabdomiosarcoma, se relaciona con su edad, sitio de origen, resecabilidad, presencia de metástasis, número de sitios metastásicos o los tejidos implicados, presencia o ausencia de participación ganglionar, el grado e histopatología de la enfermedad10,11,15-20 y las características biológicas únicas de las células tumorales del rabdomiosarcoma.
Dentro de los factores pronóstico de importancia se describen los siguientes: los niños entre uno y nueve años de edad, tienen la mejor tasa de supervivencia general.16 Los sitios primarios con un pronóstico más favorable incluyen la órbita, la cabeza y el cuello no parameníngeo, paratesticular y vagina (ni de la vejiga, ni de la próstata genitourinaria) y el tracto biliar,10,11,19,20 la carga tumoral al momento del diagnóstico tiene importancia pronostica. Los pacientes con tumores más pequeños (menores de 5 cm) tienen una supervivencia mayor comparada con niños con tumores más grandes; los niños con enfermedad metastásica en el diagnóstico tienen el pronóstico más precario.7,21 La importancia pronóstica de la enfermedad metastásica es modificada por la histología del tumor (embrionario es más favorable que otras histologías) e igualmente, el número de sitios metastásicos.17 Los pacientes con enfermedad metastásica y con tumores genitourinarios primarios (que no sean de la vejiga o próstata) tienen un resultado más favorable que los pacientes con enfermedad metastásica y tumores primarios en otros lugares;22 además, los pacientes que de otra manera muestran enfermedad localizada pero con un demostrado compromiso de los ganglios linfáticos regionales, tienen un pronóstico más precario que los pacientes sin compromiso de los ganglios regionales.20 El grado de extensión de la enfermedad después del procedimiento quirúrgico primario (es decir, el Grupo clínico), también está correlacionado con el resultado;11 en el estudio III del Intergroup Rhabdomyosarcoma Study (IRS-III), los pacientes con enfermedad residual macroscópica después de la cirugía inicial (Grupo clínico III) tuvieron tasas de supervivencia a cinco años de aproximadamente 70% comparado con una tasa de supervivencia a cinco años mayor de 90% en pacientes que no tuvieron tumor residual después de la cirugía (Grupo clínico I) y una tasa de supervivencia a cinco años de aproximadamente 80% para pacientes que presentaron tumor residual microscópico después de la cirugía (Grupo clínico II).11,15 El subtipo alveolar prevalece más entre los pacientes con características clínicas menos favorables (por ejemplo, menos de un año o más de 10 años, extremidades primarias, y enfermedad metastásica) y generalmente está relacionado con el más precario de los resultados. En los estudios IRS-I e IRS-II, el subtipo alveolar se asoció con un resultado menos favorable aun en los pacientes cuyo tumor primario fue completamente resecado (Grupo clínico I),7 sin embargo, no se observaron diferencias estadísticamente significativas de supervivencia para el subtipo histopatológico cuando se analizaron todos los pacientes con rabdomiosarcoma.23,24 En el estudio IRSIII, el resultado para pacientes con tumores del Grupo clínico I y subtipo alveolar fue similar al de otros pacientes con tumores del Grupo clínico I, pero los pacientes con subtipo alveolar recibieron terapia más intensiva.7
Clasificación histológica: El RMS se encuentra dentro de la categoría de tumores de células pequeñas redondas y azules de la infancia, el papel del patólogo es el de identificar datos característicos de linaje miogénico esquelético tanto por microscopía de luz y electrónica, inmunohistoquímica, así como biología molecular. Identificar la presencia de rabdomioblastos característicos o estriaciones de músculo esquelético por microscopía de luz, de proteínas de músculo esquelético como desmina, actina, mioglobina, proteína banda-Z, miosina y MyoD por inmunohistoquímica son indispensables para clasificarlo como RMS.25,26 El RMS varía ampliamente en su apariencia histológica, dependiendo del grado de diferenciación celular, extensión de la celularidad y patrón de crecimiento, la mayoría de estos tumores pueden clasificarse dentro de una de las siguientes cuatro categorías: embrionario, botrioides (un subtipo de embrionario), alveolar y pleomórfico, esta clasificación fue realizada por Horn y Enterline en 195827 y adoptada para la clasificación de tumores de tejidos blandos de la Organización Mundial de la Salud (OMS) en 1969. Actualizada en 1994,28 en 1995 se desarrolló la Clasificación Internacional del Rabdomiosarcoma (ICR) la cual es tanto reproducible como pronósticamente útil;29,34,36 fueron establecidos cuatro subtipos de RMS: a) RMS botrioides y de células fusiformes (10% de los casos) (variantes menos comunes de RMS embrionario), generalmente con el mejor pronóstico; b) RMS embrionario (60-70% de los casos), generalmente con un pronóstico intermedio; c) RMS alveolar (20% de los casos) (incluyendo la variante alveolar sólida), con el pronóstico más pobre; y d) sarcoma indiferenciado (pleomórfico), también con un pronóstico más pobre y casi nunca visto en niños. El rabdomiosarcoma pleomórfico se presenta principalmente en los pacientes entre 30 y 50 años de edad y difícilmente se observa en niños, en quienes el término "pleomórfico" has sido sustituido por el término "anaplásico". Finalmente, una categoría de sarcoma no específico fue creada para los tumores que no se podían clasificar dentro de algún subtipo específico.
Presentación clínica: Para fines de estudio y clasificación, se les agrupa en tumores primarios de la cabeza y cuello, que representan una proporción de 35% a 40%; de estos, 25% se desarrollan en la órbita, 50% en sitios para-meníngeos y 25% en sitios no orbitarios para-meníngeos, como el cuero cabelludo, cráneo, cara, mucosa bucal, orofaringe, laringe y cuello. Menos de 25% se origina en el tracto genitourinario y de éstos, son más frecuentes en la vejiga y próstata; aproximadamente 20% en las extremidades y el resto en primarios de tronco y otros sitios (aproximadamente 10% cada uno).9-11,30,31
Dentro de los sitios con peor pronóstico, están los tumores para-meníngeos (oído medio, senos paranasales [maxilar, etmoidal y esfenoidal], nasofaringe, fosas infratemporales, pterigopalatina y área parafaríngea), 69% de estos pacientes presentan datos de alto riesgo para el diagnóstico, como son la evidencia radiológica de extensión intracraneal, parálisis de nervios craneales y erosión de la base del cráneo.7,9,11 Otro sitio con mal pronóstico son las extremidades, asociadas a tipo histológico alveolar, diseminación linfática temprana y estadios avanzados al diagnóstico.32 Los sitios intratorácicos o retroperitoneales se presentan con una gran infiltración, poco accesibles a la resección quirúrgica completa por el involucro de vasos u órganos vitales y con una mayor probabilidad de recurrir.33
Estadificación: La evaluación de la extensión tumoral al momento del diagnóstico, es de suma importancia ya que la terapia y el pronóstico dependen del grado en el que el tumor se disemine del sitio primario. En 1972, con la formación del IRS I se generó además un sistema de estadificación clínico-quirúrgico en el que se evaluaban la resección completa del tumor, la presencia o no de enfermedad residual microscópica o macroscópica, la extensión local o a distancia del tumor así como la evidencia de metástasis.9 Esta clasificación ha sido empleada en los cuatro primero estudios realizados por el IRS (I, II y III). Otro sistema utilizado es el TNM, con base en la evaluación preoperatoria del tamaño tumoral, la presencia o no de ganglios así como de metástasis. Este sistema es ampliamente utilizado en los adultos y también es aplicado en Francia y Estados Unidos en pacientes pediátricos con sarcomas. La ventaja de esta clasificación es que no depende de factores subjetivos señalados por el cirujano, evalúa las propiedades intrínsecas del tumor, y ha demostrado un gran valor predictivo de la evolución de los pacientes;37-39 por estas razones, el IRS IV y V han empleado ambos sistemas de estadificación a partir de 1992. La identificación de variables pronósticas es de suma importancia para comprender el comportamiento de los sarcomas y el desarrollo de estudios clínicos cuidadosos, el objetivo de esto es el mejorar la sobrevida de todos los pacientes con RMS y sarcomas indiferenciados y reducir la morbilidad.40 Antes de llevarse a cabo una biopsia de masa tumoral, deben realizarse estudios de imagenología y laboratorio para sentar las bases de una evaluación. Una vez hecho el diagnóstico de rabdomiosarcoma, debe llevarse a cabo una evaluación extensiva para determinar la extensión de la enfermedad antes de empezar la terapia. Esta evaluación debe constar de rayos equis, tomografía computarizada (TC) del tórax, aspiraciones bilaterales de médula ósea y biopsias; gammagrafía ósea, imágenes por resonancia magnética de la base del cráneo y cerebro (solamente para tumores primarios meníngeos), punción lumbar para los de localización para-meníngea , así como TC del abdomen y la pelvis, en los tumores primarios de las extremidades inferiores o genitourinarias.
Tratamiento: Las tres modalidades actuales para los pacientes con sarcomas son: resección quirúrgica (si es posible), radioterapia para el control de la enfermedad residual o tumor microscópico y quimioterapia sistémica (para citoreducción primaria41,42 o erradicación de micro o macrometástasis).1 Todos los niños con rabdomiosarcoma requieren terapia de modalidad múltiple; con quimioterapia sistémica, conjuntamente con cirugía, radioterapia o ambas modalidades para el control tumoral local. Esto implica resección quirúrgica, de ser factible sin mayores trastornos de tipo funcional o cosmético, seguida de quimioterapia. Algunos pacientes con tumores inicialmente no resecados, podrían someterse a cirugía de segunda inspección para extraer residuos tumorales. Debido a que el rabdomiosarcoma es sensible a la quimio y radioterapia, la cirugía tiende a posponerse, sobre todo si contribuye a la discapacidad o interferencia con las funciones del órgano. La quimioterapia, y posiblemente la radioterapia, se administran con anticipación con la esperanza de que las resecciones quirúrgicas subsiguientes resulten exitosas sin efectos secundarios indeseables. Se recomienda la radioterapia para los pacientes con enfermedad residual microscópica (Grupo II) y enfermedad residual macroscópica (Grupo III). También se recomienda en pacientes con histología alveolar del Grupo I.43-45 De los esquemas terapéuticos para el manejo del RMS se incluyen con buena respuesta medicamentos como la vincristina (VCR), actinomicina (AMD) y cliclofosfamida (CFA) los cuales han continuando mostrando eficacia a través de los estudios realizados, han sido combinados con otras drogas como adriamicina (ADR), cisplatino (CDDP), ifosfamida (IFOS), Etopósido (VP-16), mejorando la respuesta ya alcanzada en algunos casos así como demostrando que no es necesario agregar más toxicidad para obtener respuesta en estadios tempranos con factores pronósticos favorables.7,9,11 En el estudio IRS III, fue propuesto el esquema terapéutico denominado régimen 35 para estadios avanzados II y IV, con sitio primario e histología desfavorable en el que se aplica quimioterapia neoadyuvante con 14 dosis semanales de VCR; ADR cada 10 días a partir del día 14, CDDP cada 21 días, iniciando CFA a partir del día 42; los resultados de dicho esquema fueron favorables en cuanto a la respuesta primaria del tumor así como la posibilidad de salvamento de órganos, permitiendo posponer la radioterapia hasta obtener mejor control del tumor,11 dicho régimen fue el modelo para la quimioterapia aplicada en los pacientes del INP desde el año 1993 hasta la fecha de corte del estudio.
¿ MÉTODOS
Se revisaron de forma retrospectiva y descriptiva, los expedientes de pacientes sin tratamiento previo, con diagnóstico de RMS de enero de 1997 a diciembre de 2003, tratados en el Servicio de Oncología Pediátrica del INP. Se incluyeron las siguientes variables: edad del paciente, sexo, sitio primario del RMS, tipo de cirugía inicial y grado de resección, tipo histológico, etapa clínica, presencia y sitio de enfermedad metastásica, protocolo de QT utilizado, respuesta a la QT por imagen e histopatológica, toxicidad secundaria a la QT y la evolución clínica. Se realizó un análisis univariado de estas variables y se obtuvieron curvas de sobrevida global y libre de enfermedad, por el método de Kaplan-Meier.
¿ RESULTADOS
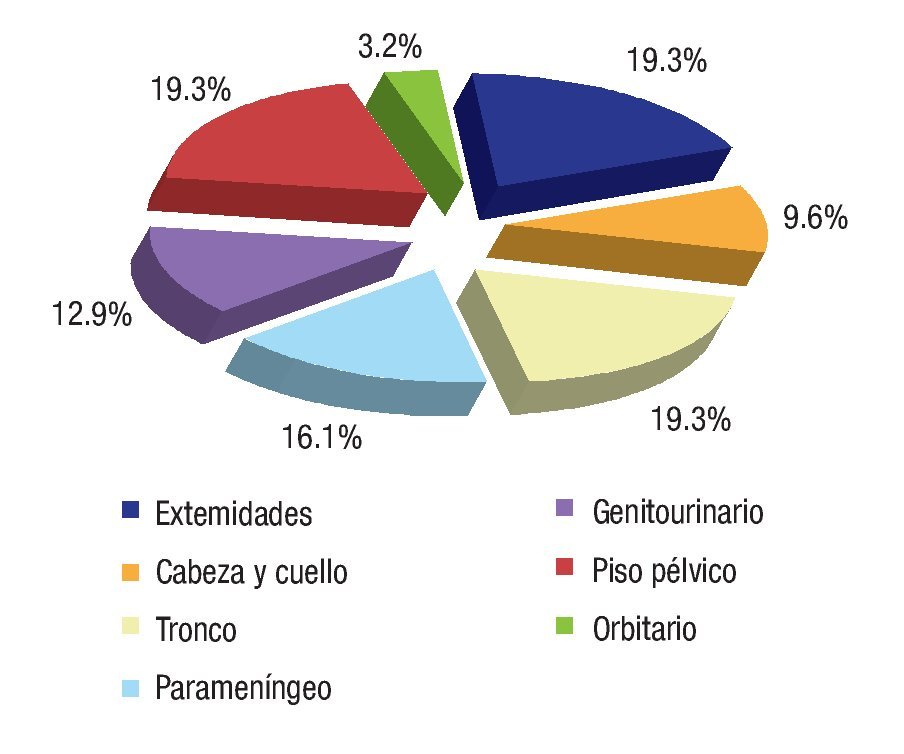
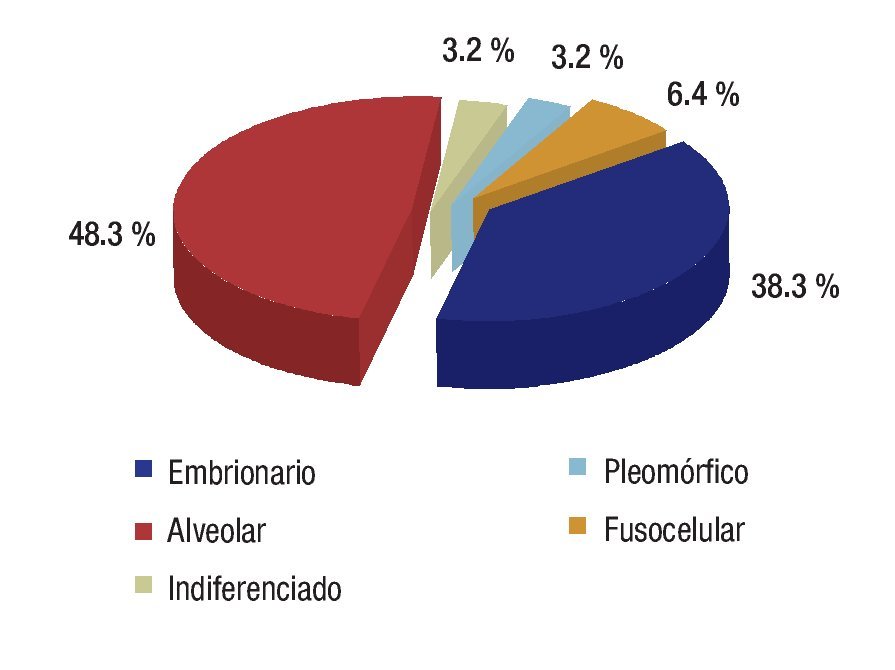
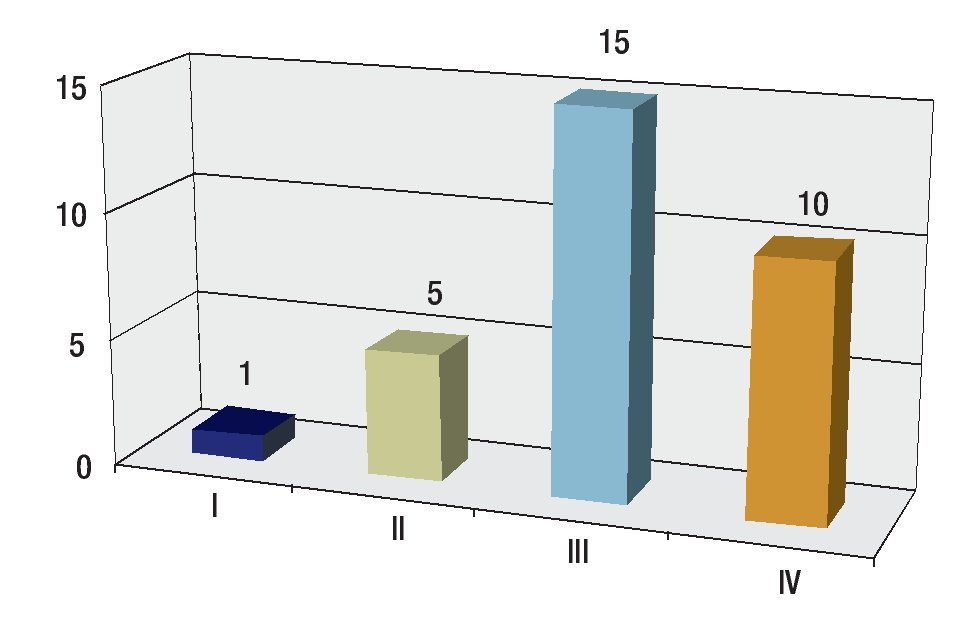
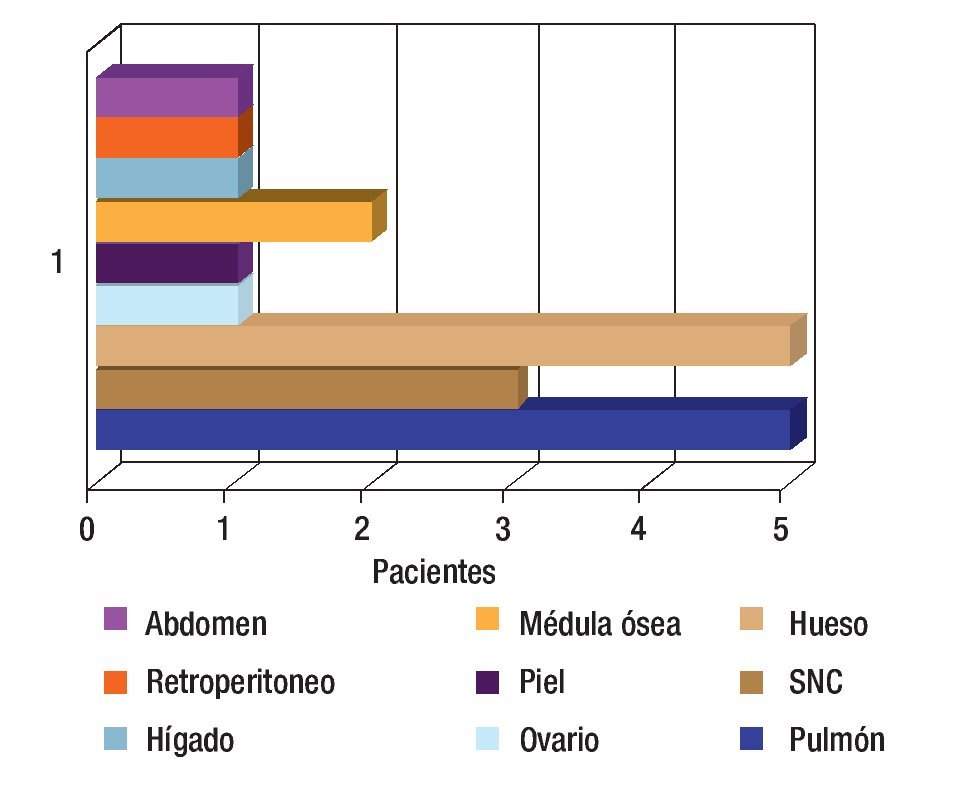
Se revisaron 35 expedientes de pacientes con diagnóstico de RMS; se excluyó a cuatro pacientes que, por abandono, no completaron la QT. Por ello, quedaron 31 casos, 11 mujeres (34.5%) y 20 hombres (64.5%); con rango de edad de 13 a 190 meses al momento del diagnóstico y mediana de 48 meses. Los sitios primarios, en orden de presentación, fueron: tronco seis (19.3%), extremidades seis (19.3%), piso pélvico seis (19.3%), para-meníngeo cinco (16.1%), genitourinario cuatro (12.9%), cabeza y cuello tres (9.6%), orbitario uno (3.2%) Figura 1; el tipo de cirugía inicial fue: sólo biopsia en 20 pacientes (64.5%), resección parcial en siete (22.5%) y resección completa en cuatro (12.9%); el tipo histológico más frecuente fue el alveolar con presentación en 15 casos (48.3%), embrionario en 12 (38.7%), fusocelular en dos (6.4%), indiferenciado en uno (3.2%) y pleomórfico en uno (3.2%) Figura 2; en cuanto al estadio los pacientes fueron evaluados utilizando en algunos ya sea el sistema TNM o la quirúrgica patológica, correspondiendo a estadios 1 (I) un solo paciente (3.2%), estadio 2 (II) cinco pacientes (16.1%), estadio 3 (III) 15 pacientes (48.3%) y estadio 4 (IV) 10 pacientes (32.2%) Figura 3; es de notar que al momento del diagnóstico 12 pacientes (38.7%) presentaron metástasis (a distancia o regional ganglionar) siendo el pulmón y hueso los sitios de mayor presentación de éstas cinco pacientes respectivamente (16.1%), seguido en frecuencia, el sistema nervioso central con tres pacientes (9.6%) y médula ósea en dos (6.4%); cinco pacientes presentaron metástasis en varios órganos (Figura 4). El tipo de QT utilizada correspondió a protocolo IRS III en 25 pacientes (80.6%) y protocolo VAI en seis pacientes (19.4%).
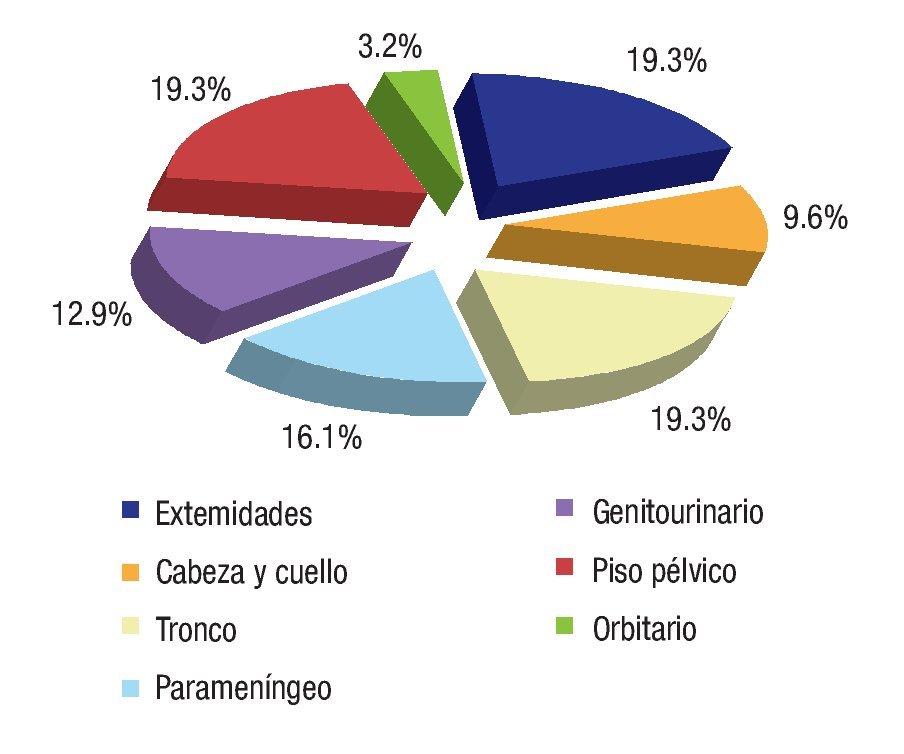
Figura 1. Sitios de presentación.
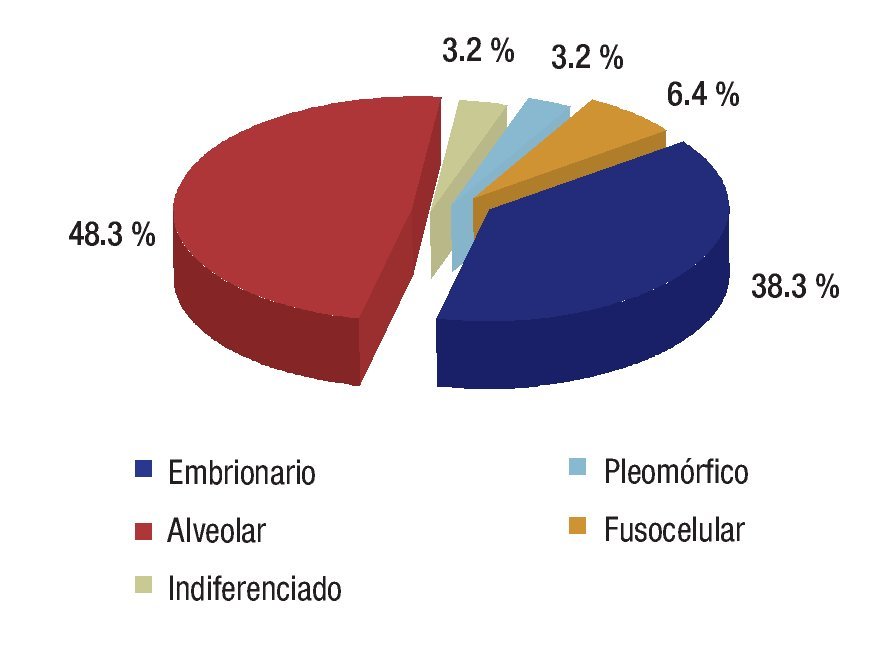
Figura 2. Tipo histológico.
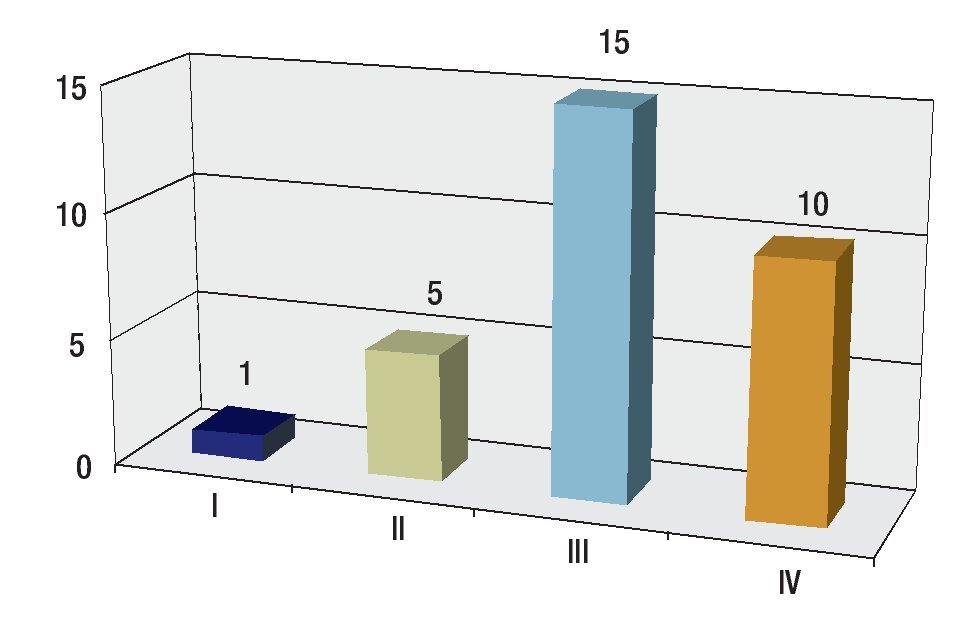
Figura 3. Distribución por estadios.
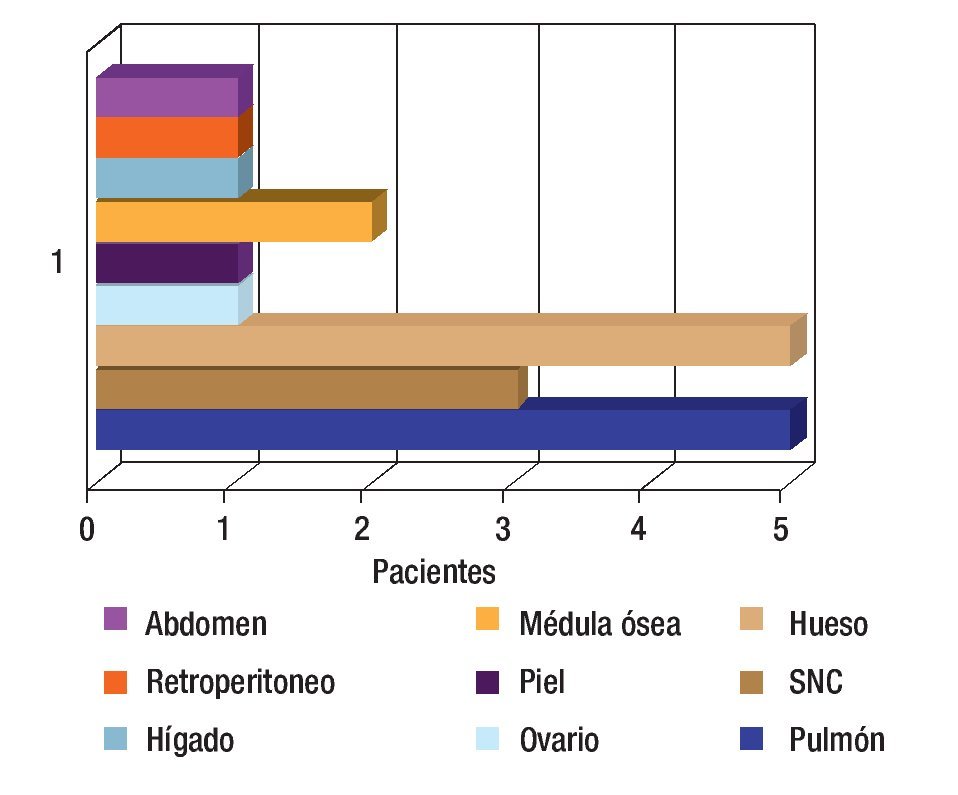
Figura 4. Sitios de metástasis.
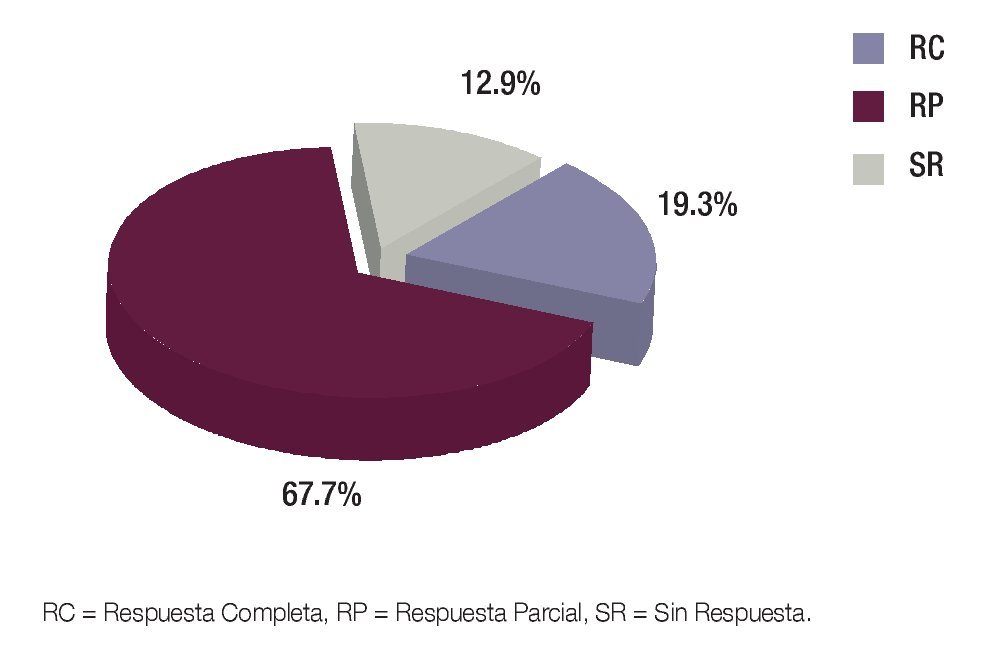
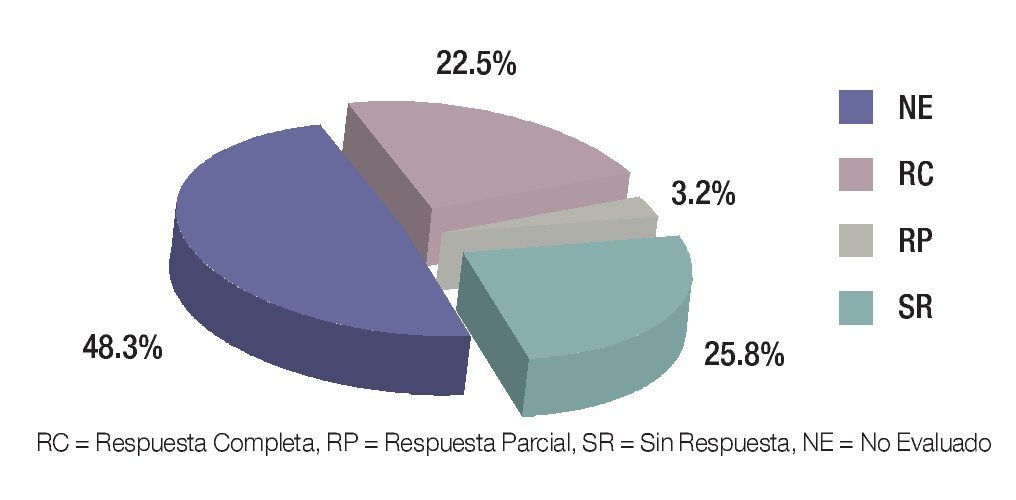
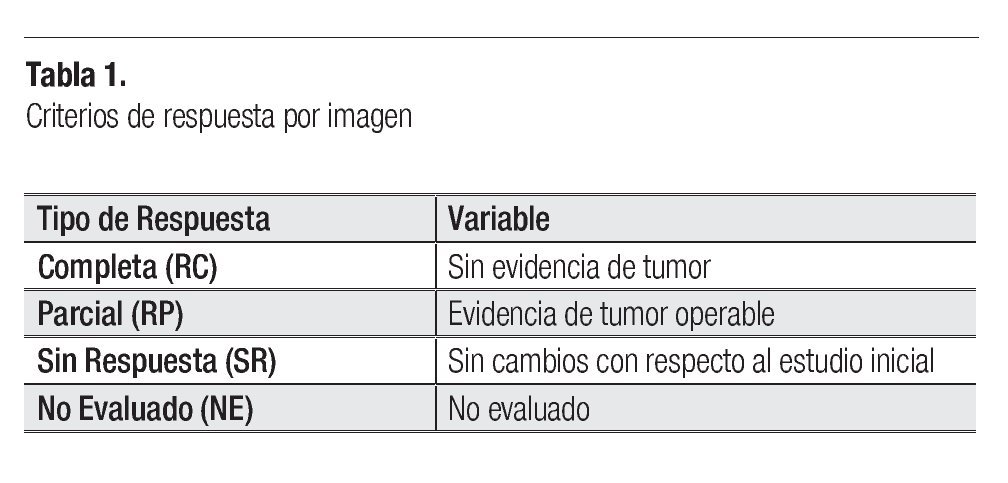
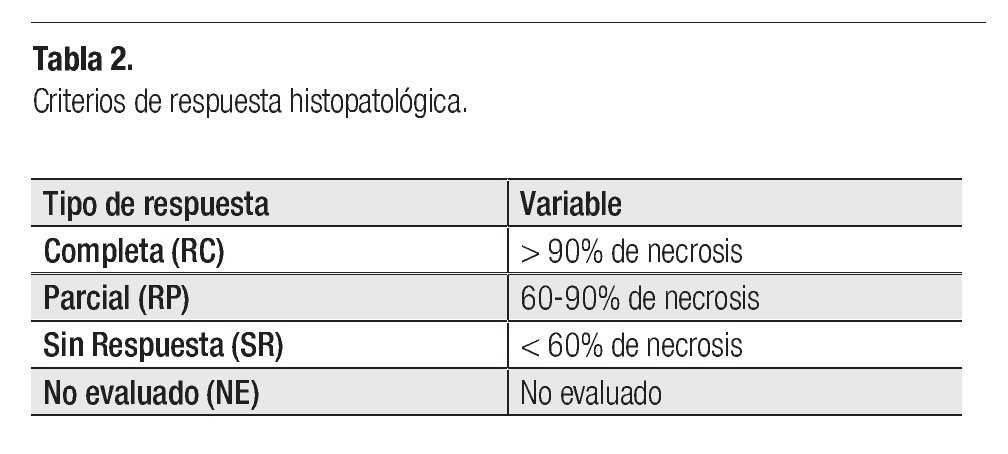
Posterior a cada ciclo de QT se realizó una evaluación clínica por imagen, tomándose en cuenta como criterios de respuesta los parámetros descritos en la Tabla 1; posterior al cuarto ciclo de quimioterapia, 21 pacientes presentaron respuesta parcial (RP) (67.7%), seis respuesta completa (RC) (19.3%) y cuatro pacientes (12.9%) sin respuesta (SR) (Figura 5). En los pacientes con respuesta completa (RC) por clínica y por imagen, se obvió la evaluación quirúrgica, ponderándose la aplicación temprana o tardía de radioterapia, dependiendo del sitio primario: en los para-meníngeos la radioterapia se administró en la semana 0, mientras que en los primarios de pelvis, alrededor de la semana 52. Los pacientes con respuesta parcial (RP) por imagen fueron sometidos a cirugía "second look" completando la respuesta con resección tumoral y evaluando el grado de necrosis y viabilidad celular. Se realizó cirugía "second look" en 23 pacientes (21 con RP y dos con SR) y de ellos, en 16 pacientes fue evaluada la respuesta histológica, donde siete tuvieron RC (22.5%), uno RP (3.2%) y ocho SR (25.8%) (Figura 6). En la Tabla 2 se exponen los criterios tomados como respuesta histopatológica.
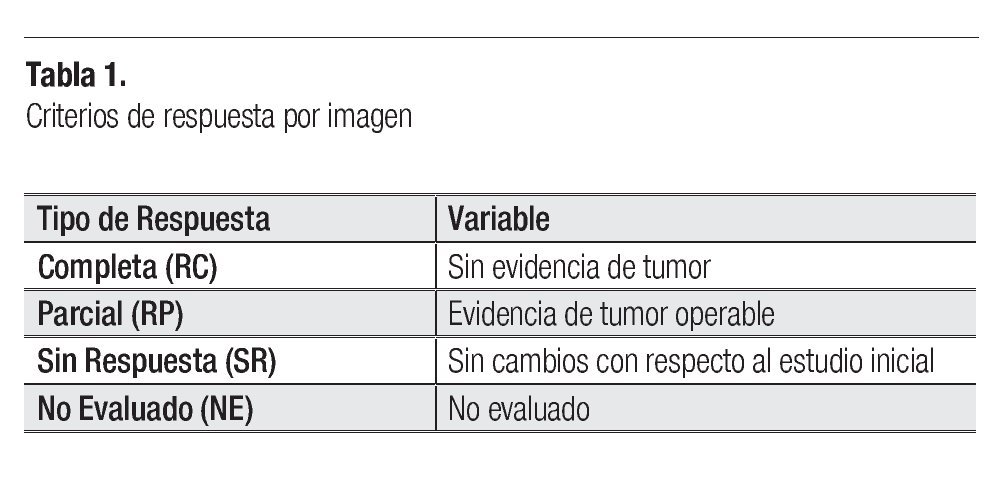
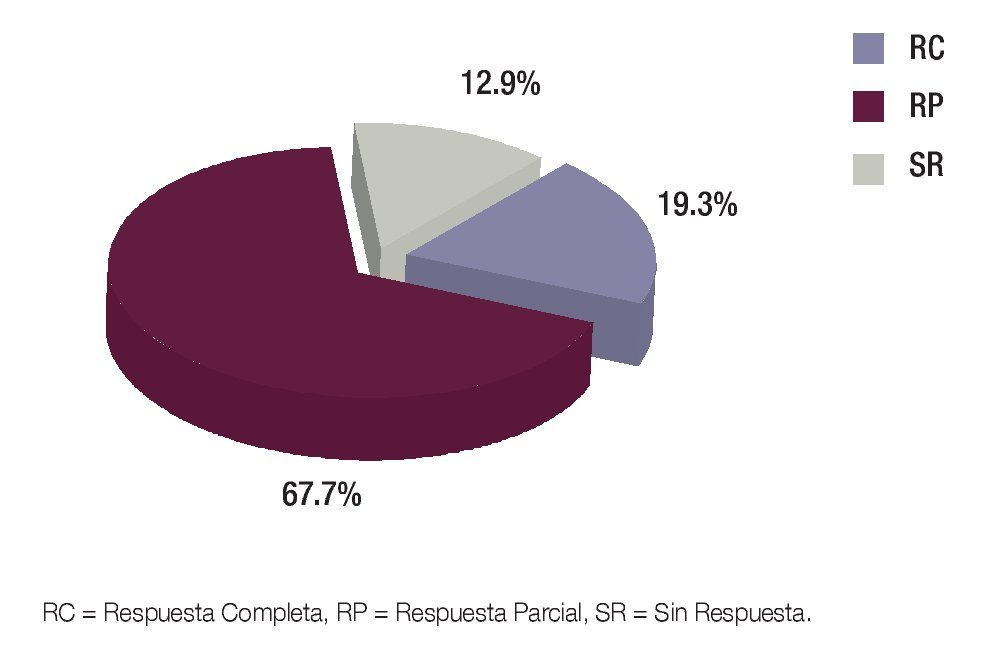
Figura 5. Tipos de respuesta por imagen.
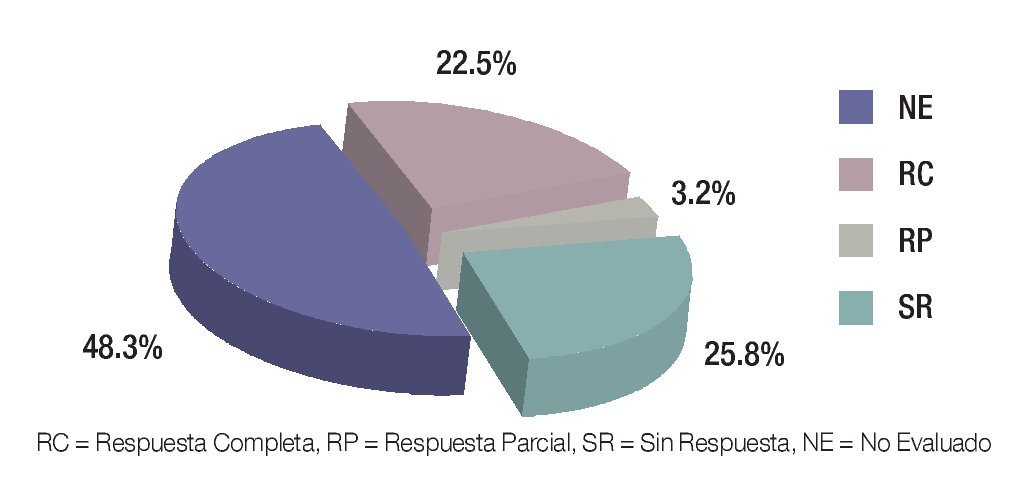
Figura 6. Tipos de respuesta por histología.
De los pacientes que por imagen tuvieron RP y en quienes no se les realizó cirugía "second look" (dos casos), uno tuvo respuesta completa posterior a la radioterapia y uno falleció por progresión de la enfermedad.
En lo que respecta a los eventos de toxicidad, la gran mayoría (28 pacientes 90.3%) tuvieron algún tipo de toxicidad, siendo la hematológica la complicación tóxica más frecuente (28 pacientes), predominando la neutropenia grado IV en 18 pacientes (58%), neutropenia grado III en nueve (29%) y neutropenia grado I en uno (3.2%); la toxicidad gastrointestinal se presentó en 12 pacientes (38.7%), presentando mucositis grado IV cuatro pacientes (12.9%), grado II y III tres pacientes respectivamente (9.6%) y colitis neutropénica dos pacientes (6.4%); 11 pacientes (35.4%) presentaron cuadros infecciosos: dos pacientes con neumonía, dos con sepsis (uno por Enterobacter cloacae), cuatro con micosis sistémica (cándida), dos gastroenteritis y uno varicela. Solamente dos pacientes (6.4%), presentaron falla renal asociada a CDDP, un paciente (3.2%) con falla hepática y otro más (3.2%) con neuropatía asociada a uso de VCR; no se presentó toxicidad a otros niveles.
Al momento de culminar el presente estudio, 16 casos (51.6%) presentaron recaída. Los sitios de presentación más frecuentes fueron: en pulmón, siete pacientes (22.5%); local, cuatro (12.9%); en abdomen dos (6.4%); un caso (3.2%) presentó segunda neoplasia un mes posterior al terminar el tratamiento (leucemia mieloide aguda).
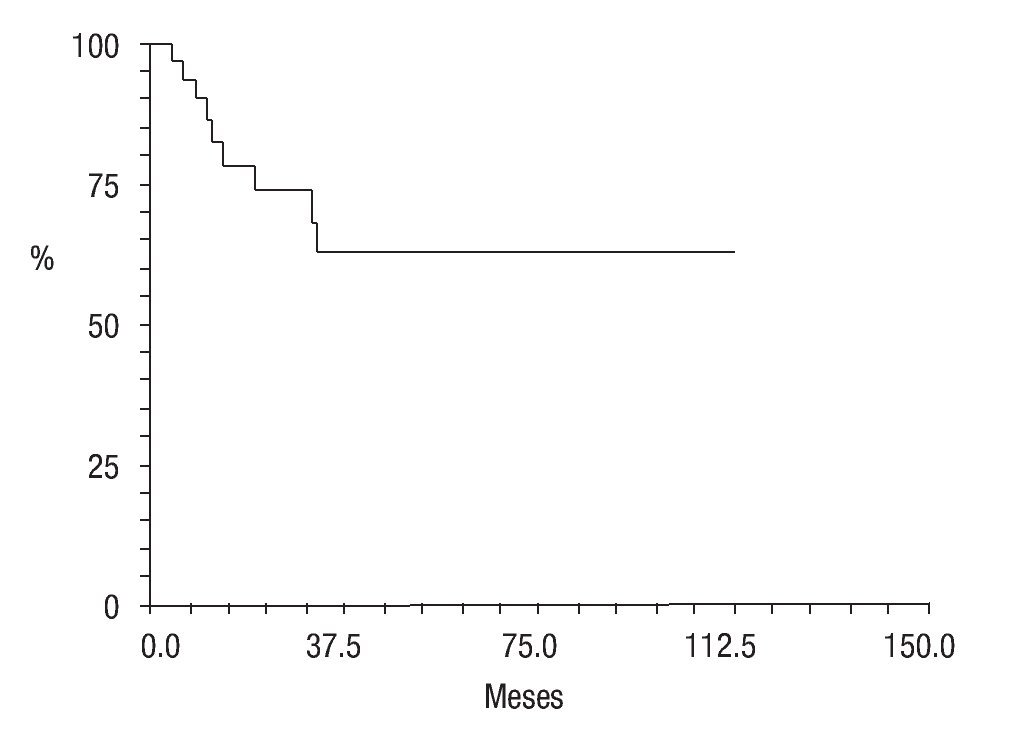
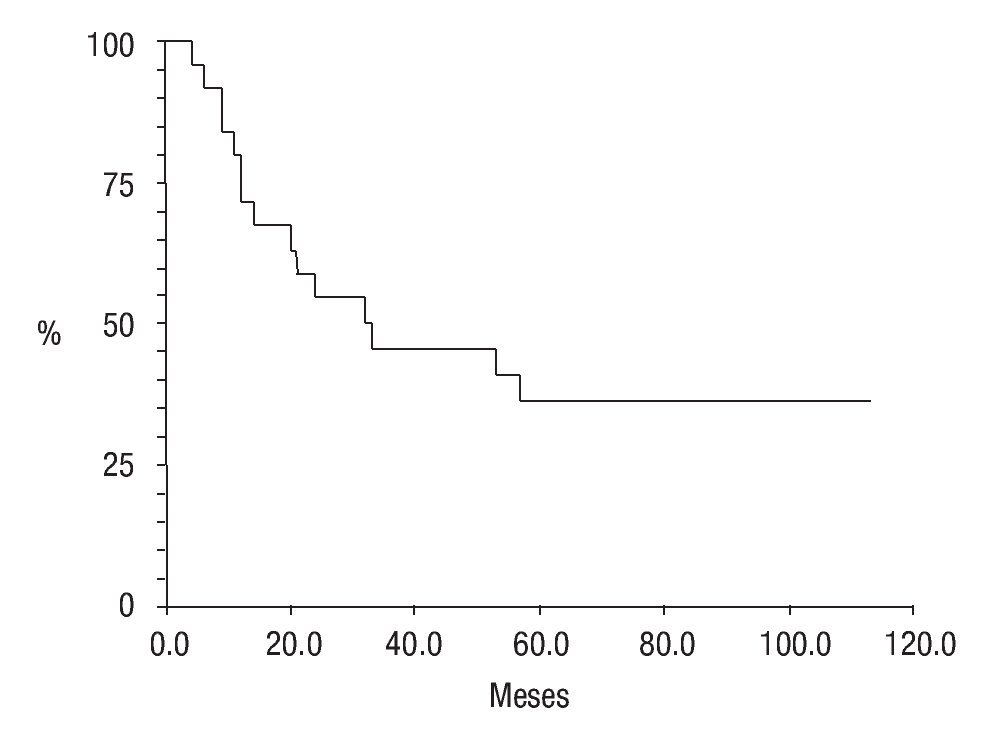
En cuanto a su estado actual, ocho pacientes (25.8%) se encontraban vivos sin actividad tumoral (VSAT); ocho vivos (25.8%) con actividad tumoral (VCAT); siete (22.5%) muertos con actividad tumoral (MCAT) y; dos (6.4%) muertos sin actividad tumoral (MSAT: uno por toxicidad hepática e infección, y otro por probable toxicidad al agente anestésico) La proporción de abandono fue de 19.2% (seis casos); la sobrevida global fue de 61% (Figura 7) y la sobrevida libre de enfermedad de 36% a cinco años de seguimiento (Figura 8).
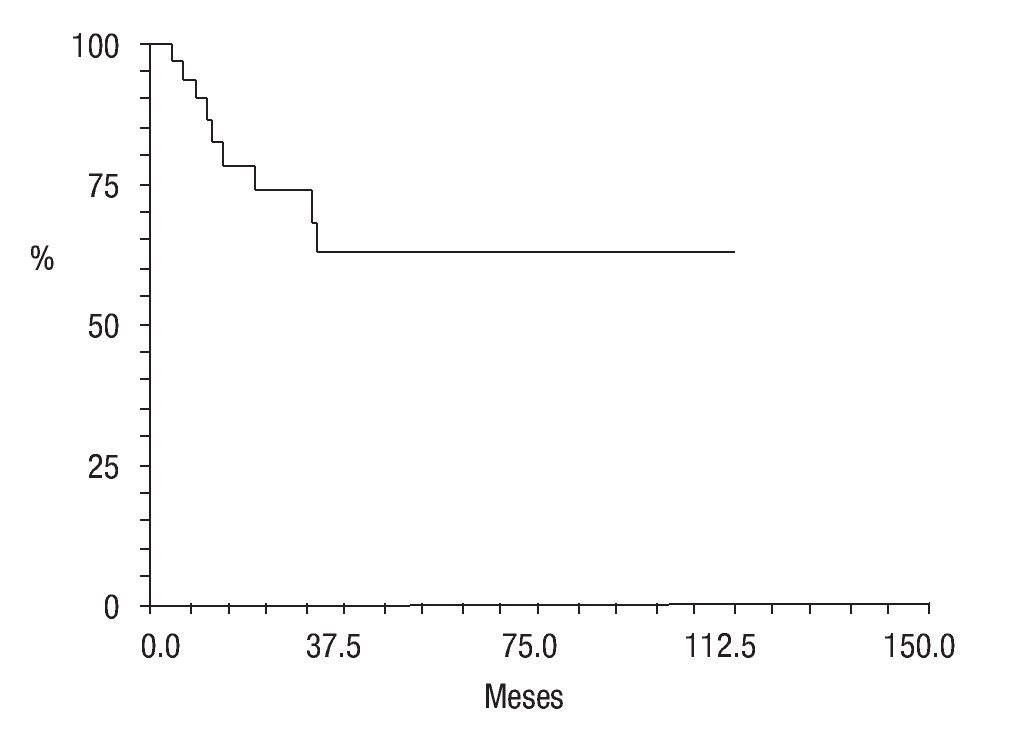
Figura 7. Sobrevida global; curva Kaplan-Meier.
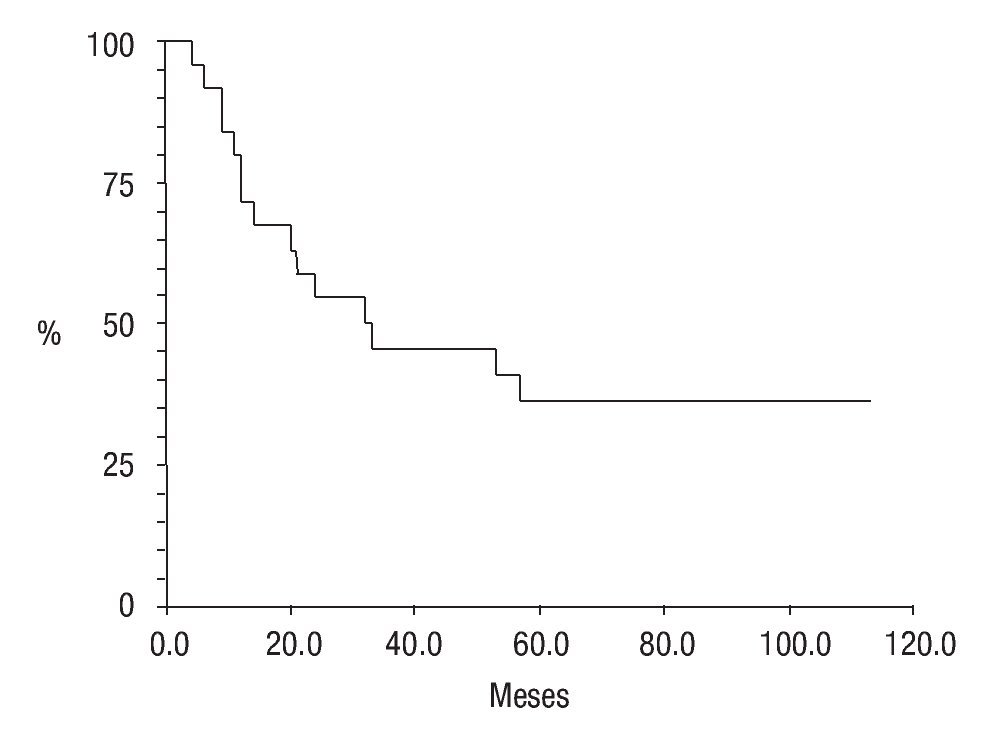
Figura 8. Sobrevida libre de enfermedad; curva Kaplan-Meier.
Al evaluar la respuesta a la QT, considerando la respuesta completa y parcial asociándola al tipo histológico, no mostró significancia estadística: embrionario (p = 0.069), alveolar (p = 0.067); ni con el estadio clínico: I (p = 0.4772), II (p = 0.1788), III (p = 0.0670), IV (p = 0.0953); ni con el sitio primario: piso pélvico, extremidades y tronco (p = 0.1904), para-meníngeo (p = 0.2301), genitourinario (p = 0.2177).
¿DISCUSIÓN Y CONCLUSIONES
El rabdomiosarcoma (RMS) es el tumor de tejidos blandos más frecuente en la edad pediátrica. En un periodo de seis años, se registraron 31 pacientes. Predominó el género masculino (64%); 80% de los pacientes se encontraban en estadio clínicos avanzados -III y IV- y el sitio de presentación que predominó fue la región de cabeza y cuello, incluidos los sitios para-meníngeos. Otros factores desfavorables fueron el tamaño tumoral (mayor a 5 cm), lo que repercutió en el control local de la enfermedad con cirugía, debido al gran volumen tumoral; la histología alveolar se presentó en casi la mitad de los casos (48%). Todos estos factores de mal pronóstico, presentes en nuestra serie, se evidenciaron en la respuesta inicial al tratamiento con quimioterapia neoadyuvante (QTN), la respuesta radiológica fue parcial en 67.7% y sólo en 19% la respuesta fue completa. En 16 pacientes se pudo realizar estudio histopatológico para evaluar la respuesta con QTN: siete pacientes (22%) tuvieron respuesta completa y en ocho casos no se obtuvo una respuesta favorable. Únicamente 25% de los pacientes estaban vivos y sin enfermedad.
En los últimos 30 años, la tasa de curación en pacientes con RMS ha mejorado en forma significativa: de entre 25% a 30%, hasta 70%. Estos resultados se atribuyen al desarrollo de protocolos internacionales de tratamiento multidisciplinario, basados en el riesgo de la enfermedad. Los principales grupos colaborativos en Norteamérica y Europa -el Intergrupo de Estudio para el RMS (IRS), actualmente conocido como el Comité de Sarcomas de Tejidos Blandos del Grupo Oncológico de Niños (COG) establecen los lineamientos de tratamiento para este tumor;9-13 en Europa, los principales grupos colaborativos (SIOP-MMT, el grupo alemán CWS y el grupo colaborativo italiano AIEOP-STSC) han unido sus esfuerzos para constituir el Grupo Europeo para el Estudio de SPB (EpSSG).44 Los mejores resultados para pacientes con RMS localizado son de 70%, pero está directamente relacionado con el grupo de riesgo. La identificación de factores pronósticos al diagnóstico ha permitido diseñar estrategias de tratamiento que han incrementado las tasas de curación aun en pacientes con enfermedad poco favorable, utilizando terapias más intensas y evitar sobre tratar y disminuir los efectos secundarios sin arriesgar la efectividad de los resultados.1
Históricamente la clasificación por estadios estaba basada en el sistema de clasificación postquirúrgica (IRS), en los que se clasificaba a los pacientes de acuerdo a la cantidad y extensión del tumor residual después de la cirugía inicial, posteriormente el IRS establece la clasificación TNM en la que se consideraban otros factores como el tamaño tumoral, la presencia de nodos y las metástasis; siguiendo estas dos clasificaciones se consideraba que la mayoría de los casos quedaban clasificados adecuadamente;37,39 sin embargo, muchos pacientes con sitios favorable podían quedar sobre clasificados o bien lo contrario, sub clasificados. En la actualidad, es necesario conjuntar estas dos clasificaciones y adaptarlas al riesgo para definir, con base en todos los factores de riesgo, el tratamiento a seguir.
La histología es una de las variables principales; la variante alveolar está asociada con un pronóstico desfavorable (61%) y 79% para RMS de otros subtipos. Estudios recientes señalan que la histología, aun siendo alveolar, no mantiene este efecto adverso en sitios favorables como el paratesticular. El sitio de origen es otro factor significativo cuando se origina en extremidades, tronco, para-meníngeos, vejiga y próstata tienen un pronóstico desfavorable, que cuando se presentan en sitios favorables como orbita, vagina, para testicular.
Por último, la edad ha sido recientemente identificada como uno de los factores pronósticos principales; los grupos de edad más desfavorables son la infancia y la adolescencia. En el estudio RMS-88 ICG, la sobrevida global fue 35% en niños menores de un año y 80% para pacientes de uno a diez años y 70% para pacientes mayores de 10 años; el IRS IV reportó una SLE de 55% para infantes, 83% para niños de uno a diez años y 68% para los mayores de 10 años; éste factor pronóstico, es independiente.12
El éxito actual para tratar niños con RMS, es combinar todos estos factores para establecer el riesgo y definir el tratamiento. El grupo Europeo Ep SSG identifica cuatro grupos de riesgo: bajo, estándar, alto y muy alto con ocho subgrupos, con recomendaciones propias para cada una de ellos. El IRS V clasifica a los pacientes con base en los mismos factores, con lo que identifica tres grupos de riesgo: bajo, intermedio y alto, con 17 subgrupos, cada uno de ellos con sus particulares recomendaciones de tratamiento.14
Es indispensable considerar los factores pronósticos para establecer los grupos de riesgo y poder mejorar la supervivencia de nuestros pacientes.
Correspondencia: Dra. Rocío Cárdenas Cardós.
Insurgentes Sur 3700-C. Col. Insurgentes Cuicuilco. Delegación Coyoacán 04530. México, D. F.
Teléfono: (55) 1084 0900, extensión 1339.
Correo electrónico:oncoped_inp@hotmail.com